Dermatologic Emergencies CME Part III: Drug reactions

Department of Dermatology, Basildon University Hospital NHS Foundation Trust, UK
Citation tools:


Copyright information
© Our Dermatology Online 2022. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
This section reviews the triad of life-threatening drug reactions including Stevens-Johnson/toxic epidermal necrolysis (SJS/TEN), drug reaction with eosinophilia and systemic symptoms (DRESS), and acute generalized exanthematous pustulosis (AGEP).
Key words: Stevens-Johnson syndrome/toxic epidermal necrolysis; Drug reaction with eosinophilia and systemic symptoms; Acute generalized exanthematous pustulosis
Severe cutaneous adverse reactions (SCARs) to drugs are potentially life-threatening and associated with various clinical patterns and morbidity. Severe cutaneous adverse reactions (SCARs) affect approximately 1 in every 1000 inpatients. Early recognition and diagnosis of SCARs are essential for improving prognosis and limiting long-term sequelae [1,2].
STEVENS-JOHNSON SYNDROME/TOXIC EPIDERMAL NECROLYSIS
Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and SJS/TEN overlap syndrome are rare mucocutaneous reactions of differing severity most commonly triggered by medications. Although the estimated incidence is low, the condition is a potentially fatal medical emergency and appropriate management is crucial for survival [3,4].
SJS/TEN represents a spectrum of reactive disorders classified according to the percentage of the affected body surface area. SJS is defined as skin involvement of < 10%, TEN is defined as skin involvement of > 30%, and SJS/TEN overlap as 10%-30%. SJS/TEN usually occurs within 7-21 days after initiation of the implicated drug. More than 100 drugs have been implicated in triggering this reaction. The most frequent culprits are sulfonamides, aromatic antiepileptic drugs, allopurinol, oxicam NSAIDs, lamotrigine, and nevirapine [5,6]. The increased risk of hypersensitivity reactions to certain drugs may be linked to specific human leukocyte antigen (HLA) alleles, immunosuppression, slow acetylation, and concomitant administration of radiotherapy and anticonvulsants [7,8].
A prodrome of fever, malaise and upper respiratory tract symptoms usually precede the eruption by several days. Erosive and hemorrhagic mucositis may result from mucous membrane involvement of the eyes, nose, mouth, and genitalia. SJS/TEN is characterized by cutaneous pain and confluent macular erythema that progresses to epidermal necrosis [9]. Extensive detachment of the epidermis sheets leads to areas of denuded oozing dermis that readily bleeds and can become secondarily infected. Septicemia is a leading cause of morbidity and mortality in the acute phase [10]. Patients surviving the acute phase of TEN have a significant risk of morbidity, including ocular lesions, cutaneous scarring, and mucosal involvement with secondary development of strictures, dental, genitourinary, gastrointestinal, respiratory, and psychological complications. The mortality rate of SJS varies between 1% and 5%, while TEN ranges from 25% to 30% [11,12].
Although a SJS/TEN diagnosis is suggested by physical signs, skin biopsy from lesional skin adjacent to a blister is usually necessary to support the clinical assessment. A second perilesional biopsy may be sent unfixed for direct immunofluorescence to exclude other types of blistering dermatoses. Epidermal necrosis seen histologically on frozen sections has high sensitivity and low specificity for the diagnosis of toxic epidermal necrolysis [13,14]. Although not readily available at this time, new experimental diagnostic tools that measure serum granulysin and high-mobility group protein B1 (HMGB1) offer the potential to differentiate early TEN from other less severe drug reactions [15,16].
A prognosis score called the Severity of Illness Score for Toxic Epidermal Necrolysis (SCORTEN) was developed to assess the severity of the disease and predict mortality in acute TEN. SCORTEN has been validated for use on days one and three of hospitalization [17] (Table 1).
 |
Table 1: Severity of illness score for toxic epidermal necrolysis (SCORTEN) [26]. |
Treatment
The mainstay of treatment for TEN involves immediate discontinuation of the culprit drug(s), specialized care in an intensive care unit or burn center, wound care, and supportive care (Fig. 1). There are no RCT data on the impact of immunomodulatory therapies in the management of TEN. The most commonly reported systemic medications that can be considered on a short-term basis include intravenous immunoglobulin (IVIG) [18], cyclosporine [19,20], pulse corticosteroids [21,22], and TNF-a inhibitors [23-25], but there is insufficient data to support their use.
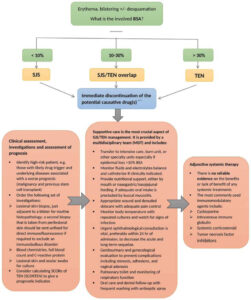 |
Figure 1: Stevens-Johnson syndrome/toxic epidermal necrolysis. Suggested algorithm for the management of SJS/TEN [27,28]. ^-Earlier withdrawal of implicated drug(s) is associated with a better overall prognosis. *-ALDEN (ALgorithm of Drug causality in Epidermal Necrolysis) is an online tool that can be used to predict the likely causality of a drug reaction. |
|
Practical pearls
|
DRUG REACTION WITH EOSINOPHILIA AND SYSTEMIC SYMPTOMS
Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, also referred to as drug-induced hypersensitivity syndrome, is potentially life-threatening, multi-organ adverse drug reaction develops 2 to 6 weeks after drug initiation (DRESS has a later onset and longer duration than other drug reactions) [31]. DRESS encompasses a combination of cutaneous rash, hematological manifestations and internal organ marked derangement [32,33]. The reaction is triggered by an interplay between predisposing drugs and reactivation of human herpesviruses (HHV), especially HHV-6, in genetically susceptible individuals [34-37]. In addition to aromatic anticonvulsant drugs, which are the most common offenders, many other drugs have been reported to trigger DRESS, including dapsone, minocycline, allopurinol, vancomycin, trimethoprim-sulfamethoxazole, abacavir, and nevirapine [38-40].
The dermatological manifestations of DRESS can be diverse, but they are most often presented as morbilliform cutaneous eruptions. The classic cutaneous distribution involves the face, upper trunk, and extremities; however, it may involve the entire surface of the skin [41]. Significant facial edema is a frequent finding that can sometimes be mistaken for angioedema [42,43].
The associated systemic involvement includes fever, lymphadenopathy, hematologic abnormalities, and multi-organ manifestations. The liver is the most frequently affected visceral organ in ~60%. 80% of patients, followed by renal, pulmonary, and cardiac manifestations. Other organs that could be involved to a lesser degree include the central nervous system, gastrointestinal and endocrine organs [44,45]. DRESS has a 10% mortality rate, most commonly from fulminant hepatitis with hepatic necrosis (Table 2).
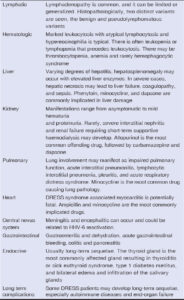 |
Table 2: Systemic complications of DRESS [49,50]. |
The diagnosis of DRESS syndrome is mainly clinical. The patient should undergo extensive evaluation to establish the diagnosis and assess the severity of internal organ involvement (Fig. 2).
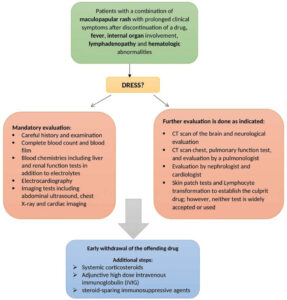 |
Figure 2: Drug reaction with eosinophilia and systemic symptoms (DRESS). Diagnostic workup for drug reaction with eosinophilia and systemic symptoms[51]. |
Treatment
Early withdrawal of the offending drug is essential, but this may not result in a rapid or complete recovery. Symptoms can take several weeks to resolve. Systemic corticosteroids show promising results for severe reactions and may help prevent autoimmune complications at the resolution stage. Adjunctive high dose intravenous immunoglobulin (IVIG) or steroid-sparing immunosuppressive agents may be used in conjunction with corticosteroids for severe and unresponsive cases of DRESS [46-48].
|
Practical pearls
|
Pitfalls in the diagnosis of DRESS
- Long latency period
- Diversity and absence of specific symptoms
- The similarity to other drug eruptions and viral exanthemas
- Delayed response after withdrawal of the culprit drug
ACUTE GENERALIZED EXANTHEMATOUS PUSTULOSIS
Acute generalized exanthematous pustulosis (AGEP) is a rare but acute severe pustular reaction that can occur within a few hours to a few days of drug exposure (earlier than other immunological drug reactions). Drugs account for approximately 90 percent of AGEP cases. The most important culprits include β-lactam antibiotics, macrolides, clindamycin, antifungals, and calcium channel blockers, especially diltiazem, but viral infections can also trigger this reaction [52-54].
AGEP is characterized by numerous pinpoints, primarily non-follicular, sterile pustules arising within large areas of edematous erythema. The rash usually begins on the face or major skin folds and rapidly extends to the trunk and limbs. Lesions typically last for 1 to 2 weeks and are followed by superficial desquamation [55,56]. In most cases, the course of AGEP is characterized by fever, leukocytosis, and elevated inflammatory markers. Systemic involvement occurs in the minority (17%-20%) of patients, primarily involving hepatic, renal, or pulmonary injury [57,58]. The overall prognosis in AGEP is good with a 1%-5% mortality rate, primarily in elderly patients and those of a poor general condition [59-61].
Patients with suspected AGEP should undergo Gram staining and pustular fluid culture to exclude potential infectious causes and the possibility of bacterial superinfection. The principal differential diagnosis of AGEP is acute generalized pustular psoriasis (Fig. 3). Lack of personal history of psoriasis, rapid appearance, shorter duration of fever and pustular eruption and recent drug exposure all favor a diagnosis of AGEP [62-64]. A skin biopsy may be warranted to distinguish the two conditions, with AGEP histopathology usually showing spongiform subcorneal and/or intraepithelial pustules, necrotic keratinocytes, an oedematous papillary dermis and perivascular infiltrate with neutrophils and some eosinophils [65].
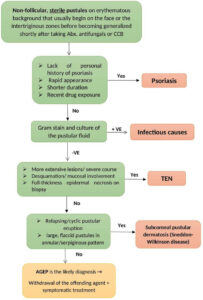 |
Figure 3: Acute generalized exanthematous pustulosis (AGEP) Flowchart of approach to generalized pustular eruption. |
Treatment
AGEP is a self-limiting disease with a favorable prognosis in most cases. Management includes removal of the offending drug, infection prevention, monitoring for systemic complications, and supportive symptomatic treatment for pruritus and fever [66,67].
|
Practical pearls
|
REFERENCES
1. Duong TA, Valeyrie-Allanore L, Wolkenstein P, Chosidow O. Severe cutaneous adverse reactions to drugs. Lancet. 2017;390:1996-2011.
2. Cho YT, Chu CY. Treatments for severe cutaneous adverse reactions. J Immunol Res. 2017;2017:1503709.
3. Patel TK, Barvaliya MJ, Sharma D, Tripathi C. A systematic review of the drug-induced Stevens-Johnson syndrome and toxic epidermal necrolysis in Indian population. Indian J Dermatol Venereol Leprol. 2013;79:389-98.
4. Frey N, Jossi J, Bodmer M, Bircher A, Jick SS, Meier CR, et al. The epidemiology of Stevens-Johnson syndrome and toxic epidermal necrolysis in the UK. J Invest Dermatol. 2017;137:1240-7.
5. Hoetzenecker W, Mehra T, Saulite I, Glatz M, Schmid-Grendelmeier P, Guenova E, et al. Toxic epidermal necrolysis. F1000Res. 2016;5:F1000 Faculty Rev-951.
6. Harris V, Jackson C, Cooper A. Review of toxic epidermal necrolysis. Int J Mol Sci. 2016;17:2135.
7. Lerch M, Mainetti C, Beretta-Piccoli BT, Harr T. Current perspectives on Stevens-Johnson syndrome and toxic epidermal necrolysis. Clin Rev Allergy Immunol. 2018;54:147-76.
8. Maverakis E, Wang EA, Shinkai K, Mahasirimongkol S, Margolis DJ, Avigan M, et al. Stevens-Johnson syndrome and toxic epidermal necrolysis standard reporting and evaluation guidelines:results of a National Institutes of Health Working Group. JAMA Dermatol. 2017;153:587-92.
9. Dodiuk-Gad RP, Chung WH, Valeyrie-Allanore L, Shear NH. Stevens-Johnson syndrome and toxic epidermal necrolysis:an update. Am J Clin Dermatol. 2015;16:475-93.
10. Lee HY, Chung WH. Toxic epidermal necrolysis:the year in review. Curr Opin Allergy Clin Immunol. 2013;13:330-6.
11. McPherson T, Exton LS, Biswas S, Creamer D, Dziewulski P, Newell L, Tabor KL, Wali GN, Walker G, Walker R, Walker S. British Association of Dermatologists’guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in children and young people, 2018. Br J Dermatol. 2019;181:37-54.
12. Hsu DY, Brieva J, Silverberg NB, Silverberg JI. Morbidity and mortality of Stevens-Johnson syndrome and toxic epidermal necrolysis in United States adults. J Invest Dermatol. 2016;136:1387-97.
13. Quinn AM, Brown K, Bonish BK, Curry J, Gordon KB, Sinacore J, et al. Uncovering histologic criteria with prognostic significance in toxic epidermal necrolysis. Arch Dermatol. 2005;141:683-7.
14. Valeyrie-Allanore L, Bastuji-Garin S, Guégan S, Ortonne N, Bagot M, Roujeau JC, et al. Prognostic value of histologic features of toxic epidermal necrolysis. J Am Acad Dermatol. 2013;68:e29-35.
15. Lim VM, Do A, Berger TG, Nguyen AH, DeWeese J, Malone JD, et al. A decade of burn unit experience with Stevens-Johnson syndrome/toxic epidermal necrolysis:clinical pathological diagnosis and risk factor awareness. Burns. 2016;42:836-43.
16. Nakajima S, Watanabe H, Tohyama M, Sugita K, Iijima M, Hashimoto K, et al. High-mobility group box 1 protein (HMGB1) as a novel diagnostic tool for toxic epidermal necrolysis and Stevens-Johnson syndrome. Arch Dermatol. 2011;147:1110-2.
17. Guégan S, Bastuji-Garin S, Poszepczynska-GuignéE, Roujeau JC, Revuz J. Performance of the SCORTEN during the first five days of hospitalization to predict the prognosis of epidermal necrolysis. J Invest Dermatol. 2006;126:272-6.
18. Tsai TY, Huang IH, Chao YC, Li H, Hsieh TS, Wang HHet al. Treating toxic epidermal necrolysis with systemic immunomodulating therapies:A systematic review and network meta-analysis. J Am Acad Dermatol. 2021;84:390-7.
19. González-Herrada C, Rodríguez-Martín S, Cachafeiro L, Lerma V, González O, Lorente JA, et al. Cyclosporine use in epidermal necrolysis is associated with an important mortality reduction:evidence from three different approaches. J Invest Dermatol. 2017;137:2092-100.
20. Roujeau JC, Mockenhaupt M, Guillaume JC, Revuz J. New evidence supporting cyclosporine efficacy in epidermal necrolysis. J Invest Dermatol. 2017;137:2047-9.
21. Law EH, Leung M. Corticosteroids in Stevens-Johnson Syndrome/toxic epidermal necrolysis:current evidence and implications for future research. Ann Pharmacother. 2015;49:335-42.
22. Liu W, Nie X, Zhang L. A retrospective analysis of Stevens-Johnson syndrome/toxic epidermal necrolysis treated with corticosteroids. Int J Dermatol. 2016;55:1408-13.
23. Woolridge KF, Boler PL, Lee BD. Tumor necrosis factor alpha inhibitors in the treatment of toxic epidermal necrolysis. Cutis. 2018;101:E15-21.
24. Scott-Lang V, Tidman M, McKay D. Toxic epidermal necrolysis in a child successfully treated with infliximab. Pediatr Dermatol. 2014;31:532-4.
25. Paradisi A, Abeni D, Bergamo F, Ricci F, Didona D, Didona B. Etanercept therapy for toxic epidermal necrolysis. J Am Acad Dermatol. 2014;71:278-83.
26. Fouchard N, Bertocchi M, Roujeau JC, Revuz J, Wolkenstein P, Bastuji-Garin S. SCORTEN:a severity-of-illness score for toxic epidermal necrolysis. J Invest Dermatol. 2000;115:149-53.
27. Schwartz RA, McDonough PH, Lee BW. Toxic epidermal necrolysis:Part II. Prognosis, sequelae, diagnosis, differential diagnosis, prevention, and treatment. J Am Acad Dermatol. 2013;69:187-e1.
28. Creamer D, Walsh SA, Dziewulski P, Exton LS, Lee HY, Dart JK, et al. UK guidelines for the management of Stevens-Johnson syndrome/toxic epidermal necrolysis in adults 2016. J Plast Reconstr Aesthet Surg. 2016;69:e119-53.
29. Zimmermann S, Sekula P, Venhoff M, Motschall E, Knaus J, Schumacher M, et al. Systemic immunomodulating therapies for Stevens-Johnson syndrome and toxic epidermal necrolysis:a systematic review and meta-analysis. JAMA dermatol. 2017;153:514-22.
30. Noe MH, Rosenbach M, Hubbard RA, Mostaghimi A, Cardones AR, Chen JK, et al. Development and validation of a risk prediction model for in-hospital mortality among patients with Stevens-Johnson syndrome/toxic epidermal necrolysis ABCD-10. JAMA Dermatol. 2019;155:448-54.
31. Camous X, Calbo S, Picard D, Musette P. Drug reaction with eosinophilia and systemic symptoms:an update on pathogenesis. Curr Opin Immunol. 2012;24:730-5.
32. Walsh SA, Creamer D. Drug reaction with eosinophilia and systemic symptoms (DRESS):a clinical update and review of current thinking. Clin Exp Dermatol. 2011;36:6-11.
33. Spriet S, Banks TA. Drug reaction with eosinophilia and systemic symptoms syndrome. Allergy Asthma Proc. 2015;36:501-5
34. Cho YT, Yang CW, Chu CY. Drug reaction with eosinophilia and systemic symptoms (DRESS):an interplay among drugs, viruses, and immune system. Int J Mol Sci. 2017;18:1243.
35. Shiohara T, Inaoka M, Kano Y. Drug-induced hypersensitivity syndrome (DIHS):a reaction induced by a complex interplay among herpesviruses and antiviral and antidrug immune responses. Allergol Int. 2006;55:1-8.
36. Miyagawa F, Nakamura Y, Miyashita K, Iioka H, Himuro Y, Ogawa K, et al. Preferential expression of CD134, an HHV-6 cellular receptor, on CD4T cells in drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic symptoms (DRESS). J Dermatol Sci. 2016;83:151-4.
37. Funck-Brentano E, Duong T, Family D, Bouaziz JD, Ortonne N, Bagot M, et al. Auto-immune thyroiditis and drug reaction with eosinophilia and systemic symptoms (DRESS) associated with HHV-6 viral reactivation. Ann Dermatol Venereol. 2011;138:580-5.
38. Kardaun SH, Sekula P, Valeyrie-Allanore L, Liss Y, Chu CY, Creamer D, et al, RegiSCAR Study Group. Drug reaction with eosinophilia and systemic symptoms (DRESS):an original multisystem adverse drug reaction. Results from the prospective R egi SCAR study. Br J Dermatol. 2013;169:1071-80.
39. Behera SK, Das S, Xavier AS, Selvarajan S. DRESS syndrome:a detailed insight. Hosp Pract. 2018;46:152-62.
40. Descamps V. DRESS syndrome. Lancet. 2022;400:560.
41. Chen YC, Chiu HC, Chu CY. Drug reaction with eosinophilia and systemic symptoms:a retrospective study of 60 cases. Arch Dermatol. 2010;146:1373-9.
42. De A, Rajagopalan M, Sarda A, Das S, Biswas P. Drug reaction with eosinophilia and systemic symptoms:an update and review of recent literature. Indian J Dermatol. 2018;63:30.
43. Choudhary S, McLeod M, Torchia D, Romanelli P. Drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome. J Clin Aesthet Dermatol. 2013;6:31.
44. Jevtic D, Dumic I, Nordin T, Singh A, Sulovic N, Radovanovic M, et al. Less known gastrointestinal manifestations of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome:a systematic review of the literature. J Clin Med. 2021;10:4287.
45. Martinez-Cabriales SA, Shear NH, Gonzalez-Moreno EI. Liver involvement in the drug reaction, eosinophilia, and systemic symptoms syndrome. World J Clin Cases. 2019;7:705.
46. Picard D, Janela B, Descamps V, D’Incan M, Courville P, Jacquot S, et al. Drug reaction with eosinophilia and systemic symptoms (DRESS):a multiorgan antiviral T cell response. Sci Transl Med. 2010;2:46ra62.
47. Shiohara T, Kano Y. Drug reaction with eosinophilia and systemic symptoms (DRESS):incidence, pathogenesis and management. Expert Opin Drug Saf. 2017;16:139-47.
48. Kim GY, Anderson KR, Davis DM, Hand JL, Tollefson MM. Drug reaction with eosinophilia and systemic symptoms (DRESS) in the pediatric population:A systematic review of the literature. J Am Acad Dermatol. 2020;83:1323-30.
49. Nguyen E, Yanes D, Imadojemu S, Kroshinsky D. Evaluation of Cyclosporine for the Treatment of DRESS Syndrome. JAMA Dermatol. 2020;156:704-6.
50. Hama N, Abe R, Gibson A, Phillips EJ. Drug-induced hypersensitivity syndrome (DIHS)/drug reaction with eosinophilia and systemic symptoms (DRESS):clinical features and pathogenesis. J Allergy Clin Immunol Pract. 2022;10:1155-67.
51. Husain Z, Reddy BY, Schwartz RA. DRESS syndrome:Part I. Clinical perspectives. J Am Acad Dermatol. 2013;68:693.e1-14
52. Sidoroff A, Halevy S, Bavinck JN, Vaillant L, Roujeau JC. Acute generalized exanthematous pustulosis (AGEP) a clinical reaction pattern. J Cutan Pathol. 2001;28:113-9.
53. Beylot C, Doutre MS, Beylot-Barry M. Acute generalized exanthematous pustulosis. Semin Cutan Med Surg. 1996;15:244-9.
54. Halevy S. Acute generalized exanthematous pustulosis. Curr Opin Allergy Clin Immunol. 2009;9:322-8.
55. Roujeau JC, Bioulac-Sage P, Bourseau C, Guillaume JC, Bernard P, Lok C, et al. Acute generalized exanthematous pustulosis:analysis of 63 cases. Arch Dermatol. 1991;127:1333-8.
56. Sidoroff A, Dunant A, Viboud C, Halevy S, Bavinck JB, Naldi L, et al. Risk factors for acute generalized exanthematous pustulosis (AGEP) results of a multinational case control study (EuroSCAR). Br J Dermatol. 2007;157:989-96.
57. Hotz C, Valeyrie-Allanore L, Haddad C, Bouvresse S, Ortonne N, Duong TA, et al. Systemic involvement of acute generalized exanthematous pustulosis:a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-32.
58. Alniemi DT, Wetter DA, Bridges AG, el Azhary RA, Davis MD, Camilleri MJ, et al. Acute generalized exanthematous pustulosis:clinical characteristics, etiologic associations, treatments, and outcomes in a series of 28 patients at Mayo Clinic, 1996-2013. Int J Dermatol. 2017;56:405-14.
59. Smeets TJ, Jessurun N, Härmark L, Kardaun SH. Clindamycin-induced acute generalised exanthematous pustulosis:five cases and a review of the literature. Neth J Med. 2016;74:421-8.
60. Fernando SL. Acute generalised exanthematous pustulosis. Australas J Dermatol. 2012;53:87-92.
61. Yek C, Gupta A, Mauskar M. Fever and a pustular rash. Jama. 2017;317:637-8.
62. Hoegler KM, John AM, Handler MZ, Schwartz RA. Generalized pustular psoriasis:a review and update on treatment. J Eur Acad Dermatol Venereol. 2018;32:1645-51.
63. Ly K, Beck KM, Smith MP, Thibodeaux Q, Bhutani T. Diagnosis and screening of patients with generalized pustular psoriasis. Psoriasis (Auckl). 2019;9:37-42.
64. Feldmeyer L, Heidemeyer K, Yawalkar N. Acute generalized exanthematous pustulosis:pathogenesis, genetic background, clinical variants and therapy. Int J Mol Sci. 2016;17:1214.
65. Kardaun SH, Kuiper H, Fidler V, Jonkman MF. The histopathological spectrum of acute generalized exanthematous pustulosis (AGEP) and its differentiation from generalized pustular psoriasis. J Cutan Pathol. 2010;37:1220-9.
66. De A, Das S, Sarda A, Pal D, Biswas P. Acute generalised exanthematous pustulosis:an update. Indian J Dermatol. 2018;63:22.
67. Szatkowski J, Schwartz RA. Acute generalized exanthematous pustulosis (AGEP):a review and update. J Am Acad Dermatol. 2015;73:843-8.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0001-6330-2856 http://orcid.org/0000-0001-6330-2856 |
