A rare case of Vohwinkel’s syndrome
Swetha Sridhar , Aneesh Samayam, Anjan Kumar Patra
, Aneesh Samayam, Anjan Kumar Patra
Department of Dermatology, MVJ Medical College and Research Hospital, Bangalore, India
Corresponding author: Dr. Swetha Sridhar
Submission: 12.07.2020; Acceptance: 13.09.2020
DOI: 10.7241/ourd.2020e.183
Cite this article: Sridhar S, Samayam A, Patra AK. A rare case of Vohwinkel’s syndrome. Our Dermatol Online. 2020;11(e):e183.1-e183.3.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Keratoderma hereditarium mutilans or Vohwinkel’s syndrome is a rare autosomal dominant type of palmoplantar keratoderma characterized by honeycomb appearance, pseudoainhum leading to auto-amputation and stellate keratosis on knuckles. It’s mode of inheritance is autosomal dominant with mutation in loricrin and connexin 26 genes. We report the case of a 35-year-old female with alopecia universalis since birth and transgradient palmoplantar keratoderma, impaired hearing and pseudoainhum formation since childhood. This case is being reported to highlight the association of Vohwinkel’s syndrome with sensorineural hearing loss and alopecia universalis.
Key words: Vohwinkel’s syndrome; Palmoplantar keratoderma; Pseudoainhum
INTRODUCTION
Hereditary palmoplantar keratodermas (PPKs) are a group of heterogeneous skin disorders which are classified based upon their mode of inheritance, age of onset, morphology and distribution of palmoplantar thickening, presence of skin lesions elsewhere, histopathology and prognosis [1]. Vohwinkel syndrome also known as keratoderma hereditaria mutilans, is a syndromic form of diffuse palmoplantar keratoderma (PPK). It is a rare autosomal dominant palmoplantar keratoderma [2]. It has been reported sparingly from all over the world [3]. It is characterized classically by a triad of diffuse hyperkeratosis of the palm and soles with honeycombing during infancy and becoming transgradient during childhood; star-shaped hyperkeratosis on the dorsa of hands, feet, knees and elbows; and pseudoainhum formation. Associated features like alopecia, deafness, spastic paraplegia, myopathy, ichthyosiform dermatoses and nail abnormalities may be noted [1].
Here we report a case of a 35-year-old female with palmoplantar keratoderma, alopecia universalis and sensorineural deafness.
CASE REPORT
A 35-year-old female born of 2nd degree consanguineous marriage presented with alopecia universalis since birth, impaired hearing and persistent palmoplantar keratoderma since childhood. No other family members had similar complaints. Palmoplantar keratoderma progressed steadily, and there was the development of fibrotic bands around fingers since the age of 25 years. There was no history of recurrent infections, teeth loss, photosensitivity, photophobia, sweating abnormalities, muscle weakness or walking disability. Rest of the systemic enquiry was also normal.
Cutaneous examination revealed well defined transgradient hyperkeratotic plaques suggestive of palmoplantar keratoderma and the presence of multiple well defined hyperkeratotic papules and plaques which were seen on the dorsum of the feet and hands (Fig. 1).
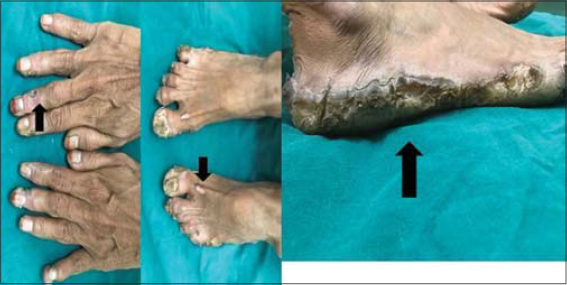 |
Figure 1. Well-defined transgradient palmoplantar keratoderma with multiple hyperkeratotic papules and plaques over dorsum of feet and hands. (black arrow – Transgradient palmoplantar keratoderma). |
Fibrotic bands were seen in the left index and little finger, suggestive of pseudoainhum (Fig. 2).
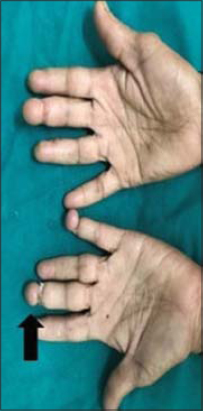 |
Figure 2. Pseudoainhum formation in left index and little finger. (black arrow – Pseudoainhum). |
Non scarring alopecia was seen involving scalp, eyebrows, eyelashes with sparse hair growth in axillary and pubic pseudoainhum (Fig. 2).
Examination of mucosa and teeth was normal.
Examination of nails revealed onychodystrophy and subungual hyperkeratosis involving the toenails.
Complete hemogram, liver function test, renal function test, urine analysis and fasting lipid profile were found to be within normal limits.
The histopathological findings from the palm consisted of marked hyperkeratosis, acanthosis, and hypergranulosis (Fig. 4).
 |
Figure 3. Non scarring alopecia over scalp, eyebrows and eyelashes. (black arrow – Non scarring alopecia). |
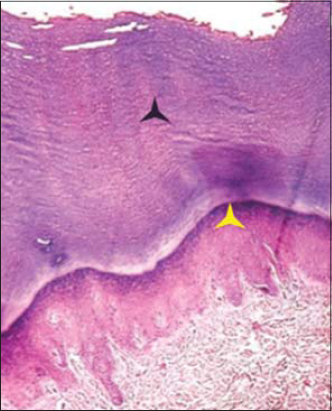 |
Figure 4. Epidermis showing hyperkeratosis, acanthosis and hypergranulosis. (black star – Hyperkeratosis) (yellow star – Hypergranulosis). |
Pure tone audiometry showed mild sensorineural hearing loss in both ears (20-40 dB HL).
Genetic analysis was not done because of financial constraints.
Patient was advised topical keratolytics and systemic retinoids but she was lost for follow-up.
DISCUSSION
Hereditary palmoplantar keratodermas (PPK) are a heterogeneous group of disorders of keratinization, the hallmark of which is a diffuse or localized thickening of palms and soles. The molecular defects in all of them have not been elaborated and these are differentiated on the basis of distribution of involvement (diffuse/insular/punctate), associated systemic features, mode of inheritance, age of onset, histopathological features, response to treatment and prognosis [4]. In 1929, Vohwinkel first described this syndrome in a 24-year-old woman who had a diffuse honeycombed palmar and plantar keratosis, in addition to distal interphalangeal constrictions since 2 years of age. The constrictions ultimately led to auto-amputation. The daughter of this patient also experienced similar clinical lesions. The patient also had mild-to-moderate sensorineural deafness [5]. Vohwinkel’s syndrome which is also known as “keratoderma hereditaria mutilans” and “mutilating palmoplantar keratoderma” is a rare autosomal dominant disorder although few sporadic cases have also been described. No racial and sex predominance is reported. The lesions start appearing in infancy or early childhood and gradually progress with age [3].
Two types of mutations of epidermal differentiation have been identified in Vohwinkel syndrome.
One is a novel missense mutation of the GJB2 gene (gap junction protein beta 2) coding connexin-26, a gap junction protein. This mutation is associated with classic, sensorineural hearing loss-associated Vohwinkel syndrome.
Another mutation is an insertional mutation of loricrin gene on the epidermal differentiation complex on 1q21. A phenotype associated with ichthyosis but not deafness is observed.
An ichthyotic variant with a 730insG mutation has also recently been described.
A case report described a new variant of Vohwinkel syndrome with congenital hypotrichosis in two siblings. A novel missense mutation in GJB2, p.Tyr65His is found to be associated with severe Vohwinkel syndrome [5].
Differential diagnosis of Vohwinkel syndrome includes acral keratoderma and Olmsted syndrome. Acral keratoderma can be differentiated by the absence of star fish shaped keratosis, presence of striate hyperkeratosis of palms, and linear hyperkeratosis over acral areas. Olmsted syndrome can be differentiated by the presence of mutilating PPK with periorificial hyperkeratosis [2].
Because of the rare occurrence of this syndrome, many treatment modalities have not been described. Topical treatment is usually not sufficient. Keratolytics like salicylates, urea and topical retinoids can reduce keratoderma. Etretinate was first used for Vohwinkel’s syndrome in 1981. Systemic retinoids can reverse keratoderma and pseudoainhum; however, relapse can occur on discontinuation of the drug [6,7].
Surgical release of the constriction bands in the form of z-plasty or cross finger flap have been described to preserve the digits [8].
CONCLUSION
This case is being reported to highlight the association of Vohwinkel’s syndrome with sensorineural hearing loss and alopecia universalis.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Ramachandra BV, Rao KP, Hithyshi V, Jhansi LS. Focal palmoplantar keratoderma with mutilating arthropathy:An interesting case report. J NTR Univ Health Sci 2014;3:114-7.
2. Mercy P, Singh A, Ghorpade AK, Das MN, Upadhyay A, Keswani N. Vohwinkel syndrome with mental retardation. Indian J Dermatol Venereol Leprol 2013;79:725.
3. Rambhia SH, Raj N, Madan MS, Prakash B. Keratoderma hereditarium mutilans (Vohwinkel’s syndrome). Indian J Paediatr Dermatol 2015;16:108-9.
4. Rani Zahida, Ahmad TJ, Hussain Ijaz. Vohwinkel’s syndrome:Case report and review of literature. Journal of Pakistan Association of Dermatologists 2003;13:92-96.
5. Choudary Nidhi, Ahar Rahul, De Abhishek, Biswas Projna. Vohwinkel’s syndrome:A rare disorder of keratinisation. Indian Journal of Dermatology 2014;59(6) :619-621
6. Camisa C, Rossana C. Variant of keratoderma hereditaria mutilans (Vohwinkel’s syndrome). Treatment with orally administered isotretinoin. Arch Dermatol 1984;120:1323-8.
7. Wereide K. Mutilating palmoplantar keratoderma successfully treated with etretinate. Acta Derm Venereol 1984;64:566-9.
8. Bassetto F, Tiengo C, Sferrazza R, Belloni-Fortina A, Alaibac M. Vohwinkel syndrome:Treatment of pseudo-ainhum. Int J Dermatol 2010;49:79-82.
Notes
Conflict of Interest: None declared.
Source of Support: Nil.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
|

Comments are closed.