Persistent purple erythematous rash: Langerhans cell histiocytosis
Imane Kacimi Alaoui , Hanane Baybay, Sara El-Ammari, Zakia Douhi, Meryem Soughi, Sara Elloudi, Fatima-Zahra Mernissi
, Hanane Baybay, Sara El-Ammari, Zakia Douhi, Meryem Soughi, Sara Elloudi, Fatima-Zahra Mernissi
Department of Dermatology, University Hospital Hassan II, Fes, Morocco
Citation tools:


Copyright information
© Our Dermatology Online 2024. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Langerhans cell histiocytosis (LCH) is an uncommon yet serious inflammatory neoplasia that frequently affects children. Skin and bone manifestations are frequent and may be isolated or associated with visceral or systemic disorders. Diagnosis is clinicopathologic, established upon typical clinical findings and immunohistochemical and histological analyses of a biopsy taken from the lesions. Prognosis depends on onset age and the quantity of affected organs. Treatment is often necessary for LCH if systemic involvement exists. Herein, we report a rare case of multifocal LCH of late onset.
Key words: Histiocytosis, Langerhans cell histiocytosis, Inflammation, Systemic manifestations
INTRODUCTION
Langerhans cell histiocytosis (LCH) is a rare disease from the histiocytosis group whose pathogenesis is unknown [1]. It is commonly reported in children, rarely in adults [2]. It ranges from a mildly asymptomatic form to a severe form involving multiple organs and is responsible for multiple visceral failures [1,3].
Herein, we report the case of Langerhans histiocytosis, particularly of late and slow onset, poorly managed due to acute leukemia associated with a pituitary adenoma.
CASE REPORT
A 24-year-old patient was followed for one year in endocrinology for pituitary adenoma under hydrocortisone and diabetes insipidus under oral antidiabetic drugs, and in oncohematology for a suspicion of acute myeloid leukemia in front of the discovery of bi-cytopenia. She was referred to our training four months previously for pruritic liquid lesions affecting the trunk and folds. The first dermatological examination revealed infiltrating, erythematous plaques covered by translucent microbubbles located on the trunk and pubic region.
Due to the suspicion of bullous dermatosis, an initial biopsy was performed, which showed dermatitis spongiform with a negative IFD result. Topical corticosteroids were initiated.
Development was characterized by clinical deterioration after three months and the appearance of new widespread skin lesions that developed in association with deterioration in general condition and tingling. The patient was hospitalized for suspected herpetic superinfection of her underlying dermatoses. Multiple plaques were found at her admission: well-defined polycyclic margins (Fig. 1a), covering the trunk and behind the ears (Fig. 1b), inguinal and intergluteal folds, with a particular distribution of the sleeveless bikini were found (Figs. 2a and 2b).
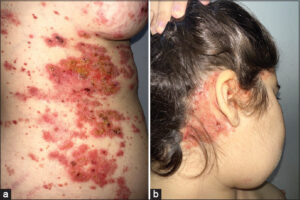 |
Figure 1: (a) Scaly, erythematous papules and plaques with polycyclic borders. (b) Seborrheic, dermatitis-like eruption. |
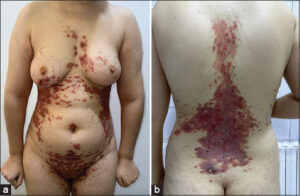 |
Figure 2: (a and b) Purpuric patches and papules taking the sleeveless bikini. |
In addition, the patient reported chronic otitis media, which was evaluated by a CT scan of the rock, which revealed mastoid filling. During his hospitalization, the patient received antiviral and antibiotic treatment with good development.
In view of this multi-lesional picture, with DS-like skin involvement involving the folds and trunk in sleeveless jersey, hematological and mastoid involvement, as well as diabetes insipidus, the diagnosis of Langerhans histiocytosis was evoked, which was confirmed by a skin biopsy with immunohistochemistry (CD1a+, CD68+, and Ps100 +, CD34-).
Her initial hematological and endocrine history was reviewed in favor of histiocytosis, and the patient was admitted to the hematology and oncology department for lesions and dilatations, showing secondary bone, liver, and lymph node locations. The patient was started on full-dose oral corticosteroids, a candidate for chemotherapy.
DISCUSSION
Langerhans histiocytosis (LH) is the most common form of histiocytosis and most commonly affects children aged 1 to 3 years, yet may occur at any age [1,3]. The male-to-female ratio is 2:1 [2]. The clinical presentations are highly variable, ranging from discrete, localized, mild, and asymptomatic forms to aggressive and widespread forms [4]. It is now more important to distinguish forms with multisystem involvement from those with localized monotissue lesions [5]. A variety of clinical manifestations are present, including punched-out lytic bone lesions, seborrheic dermatitis-like eruptions, crusted/scaly papules, plaques, and patches, eczematous lesions, diabetes insipidus, hepatosplenomegaly, cytopenia, lymphadenopathy, and a severe and fulminant multisystem condition characterized by fever, anemia, thrombocytopenia, lymphadenopathy, and hepatosplenomegaly [5–8].
Histopathological examination reveals an inflammatory infiltrate containing eosinophils, T lymphocytes, neutrophils, macrophages, and large multinucleated giant cells (LCH) [4,9].
It is important to note that LCH cells are large (10–15 cm), oval, and mononuclear. They have a complex nucleus, similar to a coffee bean, a twisted napkin, or a kidney, as well as a separate nucleolus and an eosinophilic cytoplasm. Immunohistochemistry shows positive CD1a, S100 protein, and Langerin (CD207) [1,2,4].
A thorough analytical approach, supported by the examination of the extent of the lesions, is a key element in the diagnosis of rare diseases with atypical clinical manifestations [5,10]. This was the case with our patient, who presented with systemic hematological and pituitary damage before a skin biopsy was performed, which allowed for late correction and confirmation of the LH diagnosis. Their treatment depends on the extent and location of the lesions: topical treatment (topical corticosteroids or PUVA therapy) for isolated and localized skin damage, immunosuppressants (methotrexate or thalidomide), or even chemotherapy (vinblastine and corticosteroids) for multisystem visceral damage [11,12]. Prognosis is generally poor, with systemic involvement accounting for a high mortality rate of 50–65% [13].
CONCLUSION
Langerhans histiocytosis rarely affects adults and poses a diagnostic challenge because of its variable clinical presentation [2,3]. Treatment and prognosis depend on the organs involved [12]. Skin involvement is one of the more specific symptoms of this disease, making dermatologists a crucial element in the early detection and prevention of errors in diagnosis.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Allen CE, Merad M, McClain KL. Langerhans-cell histiocytosis. N Engl J Med. 2018;30;379:856-68.
2. Kobayashi M, Ando S, Kawamata T, Makiyama J, Yokoyama K, Imai Y, et al. Clinical features and outcomes of adult Langerhans cell histiocytosis:A single-center experience. Int J Hematol. 2020;112:185-92.
3. Shukla P, Verma P, Kushwaha R, Verma S, Yadav G, Nazar AH. Langerhans cell histiocytosis in an adult female with atypical swellings. Indian J Dermatol Venereol Leprol. 2021;87:254-9.
4. Iraji F, Poostiyan N, Dehnavi PR, Soghrati M. Langerhans cell histiocytosis:A case report with unusual cutaneous manifestation. Adv Biomed Res. 2018;7:102.
5. St Claire K, Bunney R, Ashack KA, Bain M, Braniecki M, Tsoukas MM. Langerhans cell histiocytosis:A great imitator. Clin Dermatol. 2020;38:223-34.
6. Tazi A, de Margerie C, Naccache JM, et al. The natural history of adult pulmonary Langerhans cell histiocytosis:A prospective multi-centre study. Orphanet J Rare Dis. 2015;10:30.
7. Takase R, Nakano Y, Morizane S, Otsuka F. Hypothalamic mass detected in Langerhans cell histiocytosis. Intern Med. 2021;1;60:1795.
8. Zhang L, Shu H. Dermoscopic feature analysis of different skin lesions in Langerhans cell histiocytosis. Clin Exp Dermatol. 2022;47:1350-53.
9. Haroche J, Cohen-Aubart F, Rollins BJ, Donadieu J, Charlotte F, Idbaih A, et al. Histiocytoses:Emerging neoplasia behind inflammation. Lancet Oncol. 2017;18:e113-25.
10. Krooks J, Minkov M, Weatherall AG. Langerhans cell histiocytosis in children:Diagnosis, differential diagnosis, treatment, sequelae, and standardized follow-up. J Am Acad Dermatol. 2018;78:1047-56.
11. Milne P, Bigley V, Bacon CM, Néel A, McGovern N, Bomken S, et al. Hematopoietic origin of Langerhans cell histiocytosis and Erdheim-Chester disease in adults. Blood. 2017;130:167-75.
12. Tazi A, Lorillon G, Haroche J, Neel A, Dominique S, Aouba A, et al. Vinblastine chemotherapy in adult patients with Langerhans cell histiocytosis:A multicenter retrospective study. Orphanet J Rare Dis. 2017;12:95.
13. Duan MH, Han X, Li J, Zhang W, Zhu TN, Han B, et al. Comparison of vindesine and prednisone and cyclophosphamide, etoposide, vindesine, and prednisone as first-line treatment for adult Langerhans cell histiocytosis:A single-center retrospective study. Leuk Res. 2016;42:43-6.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0003-0582-321Xhttp://orcid.org/0000-0003-3455-3810http://orcid.org/0000-0001-6330-2856http://orcid.org/0000-0002-5942-441X http://orcid.org/0000-0003-0582-321Xhttp://orcid.org/0000-0003-3455-3810http://orcid.org/0000-0001-6330-2856http://orcid.org/0000-0002-5942-441X |

Comments are closed.