Psoriasis in patient with DiGeorge syndrome
Sabrina Oujdi , Meryem Soughi, Siham Boularbah, Hanane Baybay, Sara Elloudi, Zakia Douhi, Fatima Zahra Mernissi
, Meryem Soughi, Siham Boularbah, Hanane Baybay, Sara Elloudi, Zakia Douhi, Fatima Zahra Mernissi
Department of Dermatology, University Hospital Hassan II, Morocco
Citation tools:


Copyright information
© Our Dermatology Online 2023. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
DiGeorge syndrome (DGS) is a rare congenital disorder mostly caused by defects derived mainly from the third and fourth pharyngeal pouches, with additional abnormalities possibly extending from the first to sixth pharyngeal arch and first to fifth pharyngeal pouch [1]. Typically, the heart, the parathyroids, and the thymus are involved [1,2]. Other anomaly may be associated as congenital anomaly of the larynx, speech delay, gastroesophageal reflux, cleft lip and palate, absent kidney, facial abnormalities, deafness,cranial nerve palsy and thyroid disorders. Digeorge syndrome can be either complete or incomplete depending on the presence or absence of the thymus. T-cell abnormalities in individuals with DiGeorge syndrome is quite broad. Individuals might have normal T-cell numbers, low T-cell numbers, appointed incomplete DiGeorge syndrome, or no T cells appointed complete DiGeorge syndrome [3]. Most frequent form is the incomplete form while the complete form constitutes less than 1% of the cases discriebed in the literature. Patients with complete DGS may develop oligoclonal T-cell expansion, elevated IgE levels, eosinophilia, generalized skin rash, and lymphadenopathy at some time after birth, this phenotype is known as atypical complete DGS [3].
We reported a case of a 31-year-old patient followed for DGS with gastrointestinal reflux in the gastroenterology department, and who presented pruritic scaly lesion on the scalp evolving for 9 months. Clinical examination showed a normal density of the scalp and scaly patches mainly at the level of the temporal region (Fig. 1a) with a positif methodical scraping of Brocq and Auspitz sign, the dermoscopy revealed vessels in point disposes in a homogeneous way (Fig. 1b) the rest of the cutaneous examination phanerien and mucous membrane was without anomalies which led us to make the diagnosis of psoriasis of the scalp the patient was put under dermocorticoid with good evolution.
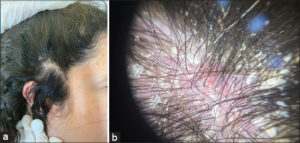 |
Figure 1: (a) Scaly patches in the scalp. (b) Vessels in point disposes in a homogeneous way on dermoscopy. |
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
REFERENCES
1. Thomas RA, Landing BH, Wells TR. Embryologic and other developmental considerations of thirty-eight possible variants of the DiGeorge Anomaly (DGA). Am J Med Genet. 1987;3(suppl):43-66.
2. Conley ME, Beckwith JB, Mancer JF, Tenckhoff L. The spectrum of the DiGeorge syndrome. J Pediatr. 1979;94:883-90.
3. Bastian J, Law S, Vogler L, Lawton A, Herrod H, Anderson S, et al. Prediction of persistent immunodeficiency in the DiGeorge anomaly J Pediatr. 1989;115:391-6.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0009-0002-5084-9152http://orcid.org/0000-0002-3975-8316http://orcid.org/0000-0003-3455-3810http://orcid.org/0000-0002-5942-441X http://orcid.org/0009-0002-5084-9152http://orcid.org/0000-0002-3975-8316http://orcid.org/0000-0003-3455-3810http://orcid.org/0000-0002-5942-441X |

Comments are closed.