Special presentation of antiphospholipid syndrome
Imane Couissi , Meryem Soughi, Kawtar El Fid, Zakia Douhi, Sara El Loudi, Hanane BayBay, Fatima Zahra Mernissi
, Meryem Soughi, Kawtar El Fid, Zakia Douhi, Sara El Loudi, Hanane BayBay, Fatima Zahra Mernissi
Department of Dermatology, University Hospital Hassan II Fès, Morocco
Citation tools:


Copyright information
© Our Dermatology Online 2023. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Antiphospholipid antibody syndrome (APAS) is a condition of acquired thrombophilia due to autoantibodies directed against membrane phospholipids and/or their cofactors. It may be primary or part of a systemic autoimmune disease, such as systemic lupus erythematosus (SLE). Dermatological lesions during APS are frequent, although non-specific, sometimes inaugural, and may be the only clinical manifestation. However, extensive cutaneous necrosis is rare and treatment is based on anticoagulants and appropriate local care. Herein, we report a case of multiple extensive cutaneous necroses in a female with SLE. The particularity of our case is the presence of two types of lesions, necrotizing plaques surmounted by hemorrhagic bullae surrounded by a purpuric border specific to antiphospholipid syndrome and ecchymotic plaques evolving according to the color of the biligenesis, which may be consistent with coagulopathy, in particular, a protein C or S deficiency, hence the interest in good knowledge and semiological analysis.
Key words: Anti-phospholipid antibody syndrome; Extensive skin necrosis; Systemic lupus erythematosus
INTRODUCTION
Antiphospholipid antibody syndrome (APAS) is a condition of acquired thrombophilia due to autoantibodies directed against membrane phospholipids and/or their cofactors characterized by venous and arterial thrombosis and/or pregnancy morbidity.
It may be primary or part of a systemic autoimmune disease, notably systemic lupus erythematosus (SLE). It affects up to 36% of patients with SLE.
Dermatologic lesions during SLE are common, although nonspecific, and may be the only clinical manifestation.
Herein, we report a case of multiple extensive cutaneous necroses in a female with SLE revealing an unknown SAPL.
CASE REPORT
We report a 39-year-old patient followed since 2012 in internal medicine for systemic lupus with hematological (anemia, lymphopenia) and cardiac (pericarditis) tropism under corticosteroid therapy with initially negative antiphospholipid antibodies. Admitted for the management of diffuse, painful, purplish-red lesions of abrupt onset evolving for one week. A dermatological examination revealed ecchymotic plaques surrounded by a purpuric halo, well-limited, with irregular contours, with the largest 10 cm in length, topped by bullae with a hemorrhagic content in some places, located on the left arm and forearm (Figs. 1a and 1b), the back, the lower abdomen, and the posterior surface of the thigh. All plaques regressed according to the biligenesis shade, except for two, which became necrotic and surrounded by a purpuric border, surmounted by hemorrhagic bullae (Figs. 2a and 2b).
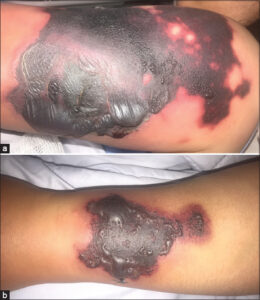
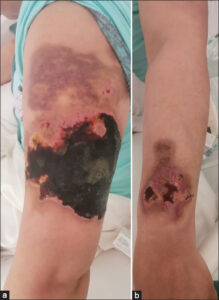
A biological workup revealed normocytic normochromic anemia at 10.9, lymphopenia at 780, thrombocytopenia at 122,000, sedimentation rate at 30 mm/1st hour, C-reactive protein at 0.05 (0.1–0.4), and normal proteins C and S.
The immunological workup was as follows: positive anti-nuclear antibodies of speckled appearance >1/160, negative anti-native DNA antibodies, positive anti-histone and anti-SSA antibodies, and positive circulating lupus-type anti-coagulant antibodies.
A skin biopsy at the level of the purpuric border revealed leukocytoclastic vasculitis with fibrinoid necrosis and the presence of micro-thrombi.
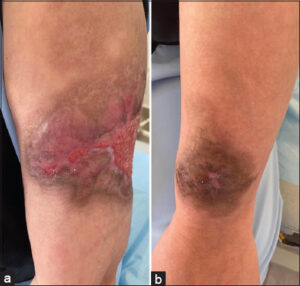
The patient was administered corticosteroids and antiplatelet agents with the regression of the ecchymotic plaques and the necrosectomy of the necrotic ones (Figs. 3a and 3b).
DISCUSSION
Antiphospholipid antibody syndrome (APAS) is a condition of acquired thrombophilia due to the presence of at least one of the circulating antiphospholipid antibodies: anti-β2glycoprotein I IgG or IgM, anti-β2glycoprotein I IgG or IgM, and/or lupus-positive circulating anticoagulants.
Dermatologic lesions during SAPL are common, although nonspecific, sometimes inaugural, and may be the only clinical manifestation [1,2].
However, extensive superficial skin necrosis remains extremely rare, reported in only 2% of cases [2,3].
The onset is brutal with necrotic purpura evolving into a blackish, escharotic plaque bordered by a purpuric border and or necrotic bullae. They are located on the limbs, face (cheeks, nose, ears), or buttocks, as in our patient.
A skin biopsy of the purpuric border shows diffuse thrombosis of the dermal and hypodermal vessels with secondary skin necrosis.
The diagnosis is based on the criteria by Myaskis et al. from 2006 [4]: the presence of at least one of the antiphospholipid antibodies and the histological confirmation of small vessel occlusion.
The particularity of our case was the presence of two types of lesions: necrotizing plaques surmounted by hemorrhagic bullae surrounded by a purpuric border specific to antiphospholipid syndrome and ecchymotic plaques evolving according to the hue of biligenesis, which may be consistent with coagulopathy, in particular a deficiency of protein C or S, hence the interest in good knowledge and semiological analysis.
The treatment is based on preventive and curative treatment with effective anticoagulation, possibly combined with corticosteroids, immunosuppressants, plasma exchange, or immunoglobulins [5–7].
Necrosectomy is essential to avoid superinfections. We must not forget to fight against other thrombotic risk factors, which are present in about 50% of patients [8] (hypertension, smoking, diabetes, obesity, vitamin D deficiency, pregnancy, postpartum, surgery, prolonged immobilization) [9].
CONCLUSION
This was a rare case of SAPL with necrotic lesions that evolved according to the color of the biligenesis, which had not yet been reported.
Hence is the interest in good semiological knowledge and analysis, which helped the diagnosis after the elimination of coagulopathies, in particular a deficiency of protein C or S.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Andrade D, Cervera R, Cohen H. Antiphospholipid syndrome:Current research highlights and clinical insights. New York:Springer. 2017:317-38.
2. Ali RA, Gandhi AA, Meng H, Yalavarthi S, Vreede AP, Estes SK, et al. Adenosine receptor agonism protects against antiphospholipid antibody-mediated NETosis and venous thrombosis. Arthritis Rheum. 2017;69:10.
3. Zuily S, Barbhaiya M, Costenbader K, Erkan D. Antiphospholipid syndrome:Current research highlights and clinical insights. New York:Springer. 2017:279-90.
4. Tedeschi SK, Johnson SR, Boumpas D, Daikh D, Dörner T, Jayne D, et al. Developing and refining new candidate criteria for systemic lupus erythematosus classification:An international collaboration. Arthritis Care Res. 2018;70:571-81.
5. Meng H, Yalavarthi S, Kanthi Y, Mazza LF, Elfline MA, Luke CE, et al. In vivo role of neutrophil extracellular traps in antiphospholipid antibody-mediated venous thrombosis. Arthritis Rheumatol. 2017;69:655-67.
6. Ünlu? O, Zuily S, Erkan D. The clinical significance of antiphospholipid antibodies in systemic lupus erythematosus. Eur J Rheumatol. 2016;3:75-84.
7. Cervera R, PintóIR, Espinosa G. Antiphospholipid syndrome:Springer. 2017:307-16.
8. Crowther MA, Legault KJ, Garcia D. Prevention and treatment of thrombotic antiphospholipid syndrome. In:Erkan D, Lockshin MD, eds. Antiphospholipid syndrome:Current research highlights and clinical insights. New York:Springer. 2017:223-34.
9. Cohen H, Hunt BJ, Efthymiou M, Arachchillage DR, Mackie IJ, Clawson S, et al. Rivaroxaban versus warfarin to treat patients with thrombotic antiphospholipid syndrome, with or without systemic lupus erythematosus (RAPS):A randomised, controlled, open-label, phase 2/3, non-inferiority trial. Lancet Haematol. 2016;3:e426-e436.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0009-0005-2261-6919http://orcid.org/0000-0002-5942-441Xhttp://orcid.org/0000-0003-3455-3810 http://orcid.org/0009-0005-2261-6919http://orcid.org/0000-0002-5942-441Xhttp://orcid.org/0000-0003-3455-3810 |

Comments are closed.