Co-existent Dowling–Degos disease, reticulate acropigmentation of Kitamura, and acropigmentation of Dohi in two generations: An overlap or concept of a single disease?
Aishwary Sabhlok , Shyam G Rathoriya, Vivek Choudhary, Rochit Singhal
, Shyam G Rathoriya, Vivek Choudhary, Rochit Singhal
Department of Dermatology, Venereology and Leprosy, Chirayu Medical College and Hospital, Bhopal, India
Citation tools:


Copyright information
© Our Dermatology Online 2023. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Reticulate pigmentary disorders (RPDs) are a group of rare autosomal dominant dermatoses with a distinctive clinical net-like pattern with specific arrangements and distributions in each entity. A forty-year-old female presented with asymptomatic, light and dark, macular lesions existing for over fifteen years with perioral scars and palmer pits and her twenty-year-old daughter beginning to develop similar lesions four years earlier. Cutaneous and histopathological examinations suggested a diagnosis of Dowling–Degos disease co-existent with reticulate acropigmentation of Kitamura in the mother and Dowling–Degos disease co-existent with acropigmentation of Dohi in the daughter. The co-existence of three infrequently encountered dermatoses with an irregular disease presentation within the family suggested the possibility of differing entities in the reticulate pigmentary group of disorders belonging to the diverse spectrums of the same disease.
Key words: Reticulate pigmentary disorders, Dowling–Degos disease, Reticulate acropigmentation of Kitamura, Reticulate acropigmentation of Dohi
INTRODUCTION
Reticulate pigmentary disorders are a group of rare pigmentary genodermatoses with autosomal dominant inheritance, comprising Dowling–Degos disease (DDD), reticulate acropigmentation of Kitamura (RAPK), and reticulate acropigmentation of Dohi (RAPD), with specific arrangements and distributions in each entity. This group of dermatoses presents clinically with freckle-like hyperpigmentation seen in inherited reticulate pigmentary disorders, while acquired disorders have the morphology of a reticulate/net-like pattern [1]. They are characterized by hyperpigmented, macular lesions with or without alternating, hypopigmented macules, pits present on the palms, soles, and perioral area, and comedo-like lesions in their various morphological subtypes. Herein, the authors describe an interesting co-existence of such lesions in a family running in two generations.
CASE REPORT
A forty-year-old, otherwise healthy, female presented with asymptomatic, light and dark, macular lesions on the dorsum of the hands, extensors of the forearms and arms, shoulders, upper trunk, neck, and face, sparing the flexors. The initial lesion appeared at the age of eighteen and progressed over fifteen years. Initially, the morphological pattern of the lesions was predominantly hyperpigmented admixed with some hypopigmented, pinhead-sized macules on the dorsum of both hands, which progressed proximally to involve the extensors of the forearms over the next five years. Further progression resulted in the involvement of the extensor aspects of the arms, nape, lateral, and frontal aspect of the neck, front, and back of the upper trunk in a sequential manner over the next ten years. The earlier lesions were light brown, with an eventual increase in pigmentation and size, as well as eruptions of new lesions noticed over the next fifteen years. The patient noticed some depressed lesions on the face and both palms with multiple, perioral, minute scars ten years into the disease onset. The lesion spread out was relatively slow in the initial five years, followed by accelerated spread out over the next decade and a halt thereafter.
Her twenty-year-old daughter began developing similar lesions four years earlier, which progressed in a similar distal-to-proximal manner with the exception of being predominantly hypopigmented and with multiple freckle-like lesions on the axilla and the absence of perioral scars and palmar pits. Both mother and daughter revealed an unequal distribution of pigmentation with intensely pigmented lesions on the face and dorsal aspects of the hands in the mother and a confluence of hyperpigmented lesions on the face in the daughter.
The patient (mother) revealed a family history of hyperpigmented, macular lesions in similar anatomical locations in her father, predicting an autosomal dominant pattern of lesions in three generations of the family. Both the patient’s and the daughter’s medical history were insignificant, and there was no history of consanguinity in the family, adverse cutaneous drug reactions, a history of chemical contact, or photosensitivity.
After obtaining written informed consent, a dermatological examination of the mother revealed numerous, widely distributed, pinpoint to pinhead-sized, predominantly hyperpigmented macules involving the dorsum of both hands and forearms, the neck, including the anterior, lateral, and nape of the neck, and upper back with pitted scars at the dorsa of the hands, as well as atrophic, hyperpigmented macules on the face, especially on the centrofacial and perioral areas (Figs. 1a – 1c). Multiple non-macular scars were also observed on both cheeks, chin, and upper lips. Multiple minute, atrophic pits were also present on both her palms (Fig. 1d).
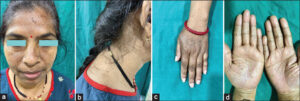
A dermatological examination of the daughter revealed predominantly hypopigmented macules presented on the dorsa of the hands, forearms, and feet with hyperpigmented macular lesions on the axillae, inner aspects of both arms, anterior, lateral and posterior part of the neck, forehead, perioral, and malar area (Fig. 2a). Interestingly, hypopigmented macules on the dorsa of the hands and feet were observed on a hyperpigmented background. No palmar pits or perioral scars were observed in the daughter. The presence of axillary, freckle-like lesions and hypopigmented macules on the dorsa of the hands and feet were the distinctive findings in the daughter from the mother (Figs. 2b – 2d). The patient’s (mother) father was unavailable for clinical examination.
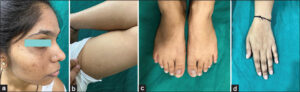 |
Figure 2: (a) Clinical photographs of the daughter showing multiple hyperpigmented macules on the face. (b) Clinical photographs of the daughter showing hyperpigmented macules on the axilla. (c) Clinical photographs of the daughter showing a hypopigmented lesion on the dorsum of feet. (d) Clinical photographs of the daughter showing a hypopigmented lesion on the dorsum of the hands. |
Both patients’ routine lab investigations, including a complete blood count, liver and kidney profile, blood glucose, ECG, chest X-ray, and peripheral smear for atypical cells, were all within normal range.
A skin biopsy from the mother, taken from a hyperpigmented macule on the upper back revealed focal thinning of the epidermis, dilated follicular infundibula with a keratin cyst, and a pigmented basal layer having one focus showing elongation of rete ridges. Except for minimal perivascular lymphocytic infiltrates, there was no evidence of pigment incontinence in the dermis (Fig. 3a).
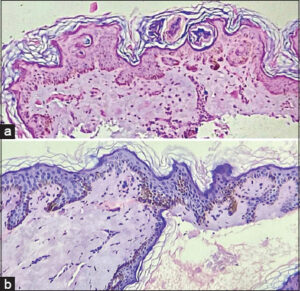 |
Figure 3: (a) Histopathological examination of the hyperpigmented lesion in the mother showing focal thinning of the epidermis, dilated follicular infundibula with a keratin cyst, and a pigmented basal layer (H&E, 40×). (b) Histopathological examination of the hyperpigmented lesion in the daughter showing flattening of rete ridges, an atrophic epidermis, and marked basal hyperpigmentation with an antler-like pattern of rete ridges without pigment incontinence (H&E, 40×). |
A skin biopsy taken from a single hyperpigmented lesion on the inner aspect of the arm of the daughter and a further histopathological examination revealed flattening of rete ridges, an atrophic epidermis, marked basal hyperpigmentation with focal areas showing a reticulate, interdigitating, antler-like pattern of rete ridges without evidence of pigment incontinence. The dermis appeared normal except for collagenization (Fig. 3b). The histomorphological impression suggested Dowling–Degos disease. Histomorphology suggested a similar impression to that of the mother.
On the bases of the history, cutaneous examination, and its histopathological correlation, the diagnosis of reticulate pigmentary dermatosis was reached with predominant features of Dowling–Degos disease co-existent with reticulate acropigmentation of Kitamura in the mother and features suggestive of Dowling–Degos disease co-existent with acropigmentation of Dohi in the daughter.
DISCUSSION
Reticulate pigmentary disorders (RPDs) are a group of rare autosomal dominant dermatoses with variable penetrance, having a specific pattern of distribution and consisting of variously named disorders, primarily Dowling–Degos disease (DDD), characterized by asymptomatic, pigmented, reticulate macules. Reticular hyperpigmentation of Kitamura (RAPK) is characterized by asymptomatic, pigmented macules on the dorsa of the hands, forearms, and feet with palmoplantar pits. Likewise, reticulate acropigmentation of Dohi (RAPD) is characterized by a combination of hypo- and hyperpigmented macules on the hands and feet.
DDD, first described by Dowling in 1938 and by Degos in 1954, often has a late-onset presentation of reticulate, pigmented lesions on flexures, that is, the axillae, groin, and neck, with comedo-like lesions and pitted perioral scars [2]. In addition, DDD has reportedly been associated with other diseases, such as hidradenitis suppurativa, multiple keratoacanthomas, and squamous cell carcinoma [3–5].
Likewise, reticulate pigmentation of Kitamura (RAPK), described by Kitamura and Akamatsu in 1943, has increasingly been reported across the world, with a presentation beginning usually in childhood or early teenage years. Lesions of RAPK are described as symmetrical, hyperpigmented macules in a reticulate pattern on the dorsa of the hands and feet, with atrophy being the characteristic feature of early lesions [6]. Progressive extension of lesions occurs proximally with age, along with pitted lesions on the palmoplantar surfaces with altered dermatoglyphics and the disruption of epidermal ridges.
Reticulate acropigmentation of Dohi (RAPD) presents as areas of hyper- and hypopigmentation on the dorsal and ventral aspects of the hands and feet, which may extend proximally to involve the knees and elbows [7]. Lesions of RAPD appear during the first decade of life and their progression decelerates by adolescence. This condition spares the mucosae and does not usually exhibit specific palmoplantar pits or perioral pitted scars. The predominance of hypopigmented macules in this subset makes it distinctive in terms of presentation, unlike other common variants of RPDS.
Our two patients (mother and daughter) exhibited interesting features of a complex hereditary disorder, with DDD and RAPK being consistent in both, yet in overlapping/co-existent patterns, as well as the evolution of early features of RAPD as hypopigmented macules in the daughter, gradually shifting to evolved features of DDD and RAPK in the form of hyperpigmented macules in mother, suggesting the distinct nature of the disease in terms of the transformation of phenotypic expression in the subsequent generation. The feature has not been reported elsewhere so far, although photo-aggravated changes and the darkening of macules of prototype APD cannot be excluded in the mother in this case.
We also observed a relatively faster rate of transition of RAPD features (hypopigmented macules to hyperpigmented macules) in the daughter, possibly explained by variable point mutations being expressed sporadically [8].
The daughter revealed hypopigmented macules on a hyperpigmented background on the dorsa of the hands, forearms, and feet (suggestive of RAPD). Some hyperpigmented macules were present on the axillae, inner aspect of the arms, and cento-facial area (suggestive of DDD), without hyperpigmented macules on the dorsa of the forearms or palmar pits, features typical of RAPK.
Similarly, pitted hyperpigmented macules chiefly on the perioral and periorbital areas, on the neck, upper back, and axillae in the mother were suggestive of DDD. Multiple pits on both palms along with hyperpigmented, atrophic macules on both forearms supported RAPK clinically, lacking morphological features of RAPD, although, earlier, the patient had hypopigmented macules on both forearms, which gradually darkened over years, chronologically suggesting the gradual transformation of one subset of RPD to another, considering the pattern and evolution of similar lesions on the forearms in the daughter at the time.
The clinical presentations of various RPDS depend upon the age at which they present, the selective predominance of specific subtypes of RPDS, and the impact of morphological variations on a selective group of populations with genotypic heterogeneity to manifest the predominant or co-existent RPDS [9].
There have been numerous case reports of the co-existence of different cases of RPDS in the literature, suggesting an overlap or a single entity with variants [10–12].
The pathogenesis of RPDS has not been understood clearly, with some studies indicating mutations in RNA-specific adenosine deaminase genes in RAPD, a mutation in the ADAM10 and PAX2 genes in RAPK and a loss of mutation in KRT5 in DDD. Wei Tai Yu et al. found an increased percentage of stage 4 melanosomes in basal and suprabasal keratinocytes in lesional skin, suggesting the involvement of defective melanosome transfer and maturation in the pathogenesis of DDD [9].
Our study found a similar overlap as described by Thami et al. in 1998, yet we found an apparent overlap in two subsequent generations with the polarization of genodermatoses in the mother and impending phenotypic expression in the daughter, making the current report interesting and unique. [10].
CONCLUSION
The possibility of variable phenotypic expressions of a single disease entity depending upon the age of presentation, site, duration, and various environmental and internal factors should be considered. The co-existence of three infrequently encountered dermatoses with an irregular disease presentation within the members of the same lineage with a lack of distinguishing histomorphological features should raise the question of diverse features of a single disease entity.
There have been numerous reports on the co-existence of RAPK and DDD in the literature, and numerous authors have suggested the possibility of differing entities in the reticulate pigmentary group of disorders belonging to the diverse spectrums of the same disease. Our report showed a non-idiosyncratic co-existence of the reticulate pigmentary group as a single entity in two generations of a family with different phenotypic outcomes, making it a thought-provoking observation.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Sardana K, Goel K, Chugh S. Reticulate pigmentary disorders. Indian J Dermatol Venereol Leprol. 2013;79:17-29.
2. Topal S, Topal IO, Bilgi H. A case of delayed diagnosis of Dowling–Degos disease. Our Dermatol Online. 2023;14:114-5.
3. Mancy A. Dowling-Degos disease:An association with hidradenitis suppurativa. Our Dermatol Online. 2022;13:445-8.
4. Fenske NA, Groover CE, Lober CW, Espinoza CG. Dowling-Degos disease, hidradenitis suppurativa, and multiple keratoacanthomas:A disorder that may be caused by a single underlying defect in pilosebaceous epithelial proliferation. J Am Acad Dermatol. 1991;24:888-92.
5. Ujihara M, Kamakura T, Ikeda M, Kodama H. Dowling–Degos disease associated with squamous cell carcinomas on the dappled pigmentation. Br J Dermatol. 2002;147:568-71.
6. Tang JC, Escandon J, Shiman M, Berman B. Presentation of reticulate acropigmentation of Kitamura and Dowling–Degos disease overlap. J Clin Aesthet Dermatol. 2012;5:41-3.
7. Sinha S, Kulhari A. Reticulate Pigmentary Disorders:A review. Pigment Int. 2019;6:67-76.
8. Li M, Yang LJ, Zhu XH, Zhang HP, Dai XY. Analysis on the mutation of ADAR gene in a pedigree with dyschromatosis symmetrical hereditaria. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 2007;24:446-8.
9. Yu Wei-Tai, Su Yung-Shun, Lee Chih-Hung. A Taiwanese woman with Dowling–Degos disease:An electron microscopic study with pathophysiological significance. Dermatol Sinica. 2014;32:33-6.
10. Thami GP, Jaswal R, Kanwar AJ, Radotra BD, Singh IP. Overlap of reticulate acropigmentation of Kitamura, acropigmentation of Dohi and Dowling Degos disease in four generations. Dermatol. 1998;196:3501.
11. Rathoriya SG, Soni SS, Asati D. Dowling–Degos disease with reticulate acropigmentation of Kitamura:Extended spectrum of a single entity. Indian Dermatol Online J. 2016;7:32-5.
12. Vasudevan B, Verma R, Badwal S, Pragasam V, Moorchung N, Badad A. A case of reticulate acropigmentation of Kitamura:Dowling–Degos disease overlap with unusual clinical manifestations. Indian J Dermatol. 2014;59:290-2.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
|

Comments are closed.