Advances in targeted strategies for managing neurofibromatosis type 1-related tumors
Zhang Li1, Rajbanshi Bhavana1, Shrestha Surendra2, Li Xiuli 3, Zhao Jingjun1
3, Zhao Jingjun1
1Department of Dermatology, Tongji Hospital, Tongji University School of Medicine, Shanghai, China, 2Emergency Department, Om Aasha Hospital Pvt. Ltd., Dhangadhi, Nepal, 3Department of Dermatology, Shanghai Tenth People’s Hospital, Tongji University School of Medicine, Shanghai, China
Citation tools:


Copyright information
© Our Dermatology Online 2023. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Neurofibromas are the most common and disfiguring feature of neurofibromatosis type 1 (NF1). The treatment options for NF1 have been limited to surgical removal, yet in some cases, the growth pattern of neurofibroma may make its complete resection unpractical. Practitioners are attempting to determine the treatment options for NF1-related tumors that may shrink tumor size, which may cause local organ compression or even decrease the potential long-term risk of undergoing malignant transformation. Several clinical trials evaluating targeted therapeutics reported to have achieved promising results, including Raf inhibitors (sorafenib), MEK inhibitors (selumetinib and trametinib), mammalian target of rapamycin (mTOR) inhibitors (rapamycin), and those targeting the tumor environment (imatinib mesylate and pirfenidone). In 2018, due to high efficacy and low side effects of selumetinib symptomatically and progressively for inoperable plexiform neurofibromas, it was granted orphan drug designation by the FDA and the European Medicine Agency. In this review, we discuss the most common types of NF1-related tumors and the possible mechanisms of tumorigenesis, including the contributions of different signaling pathways and the tumor microenvironment for its management. We also focus on the recent notable advances in the development of therapeutic strategies for NF1-related tumors, including the compounds that have completed their clinical trials and the promising drugs still in clinical trials that have not shown their outcomes to provide perspective to researchers for future studies.
Key words: Neurofibromatosis; Neurofibromas; Therapeutics; Inhibitors
INTRODUCTION
Neurofibromatosis type 1 (NF1) is an autosomal dominant and multisystem disorder that affects approx. 1 in 2500–3000 individuals worldwide [1]. The defining manifestation of NF1 is the presence of neurofibroma with a considerable variation in clinical features such as pigmentary abnormalities, skin freckling, Lisch nodules, and behavioral abnormalities [2], even among monozygotic twins. In addition, there are few correlations between genotype and phenotype, with much depending on stochastic events [3].
Recent decades have revealed the pathogenesis of NF1, which is frequently caused by the mutation of the NF1 gene, a tumor suppressor gene that resides on chromosome 17 and encodes Ras-GTPase activating a protein known as neurofibromin. It is a 2818-amino acid cytoplasmic protein that negatively regulates the Ras cascade by accelerating the conversion of Ras from the active to inactive form [4]. With its decrease, Ras signaling pathways sustain hyperactivity, subsequently leading to uncontrolled cell proliferation and differentiation. Recently, research on developing compounds aimed at the mechanism of tumorigenesis has become a hotspot. Inhibitors targeting the pathogenesis of neurofibroma showed promising clinical improvement in some clinical trials [5,6]. New therapies for NF1related tumors may be divided into those targeting the tumor microenvironment and those targeting the Ras pathways within NF1deficient tumor cells. Herein, we will focus on the pathogenesis of NF1-related neurofibromas, discuss the preclinical and clinical research accumulated over the past several years, and provide more therapeutic options for clinicians to treat NF1-related tumors.
NEUROFIBROMAS
Neurofibromas are histologically benign tumors consisting of Schwann cells, fibroblasts, mast cells, macrophages, lymphocytes, and other elements of the nerve. All neurofibromas have certain histological and cellular characteristics [7]. Two subtypes—cutaneous neurofibromas (CNFs) and plexiform neurofibromas (PNFs)—are often seen.
CNFs are tumors with the highest prevalence and almost all patients with NF1 will experience cutaneous tumors. They originate from small peripheral nerve branches and are always limited to the skin. CNFs are histologically benign tumors and have no possibility of progression to a malignant form [8]. However, they may manifest as thousands of nodules, and the number increases with increasing age. Although they are not life-threatening, they may have a significant influence on physical appearance and mental health of the patient [9].
Approx. 20–50% of patients with NF1 will develop PNFs, which may be present in various regions of the body and all stages of life. PNFs develop from cranial and large-peripheral nerve sheaths and may easily invade the adjacent tissues and occasionally result in lifelong disfigurement, pain, and mortality [10]. Furthermore, around 7–12% of patients with PNF have a lifetime risk of developing malignant peripheral nerve sheath tumors (MPNSTs) [11].
MPNSTs are highly rare soft-tissue sarcomas, with an incidence of 0.001% in the general population, yet its incidence is higher in NF1. MPNSTs arise from peripheral nerve cells and develop from PNFs or secondary to radiation therapy [12]. Their specific histogenesis is unclear. The aggressive growth pattern and chances of early metastasis make MPNSTs a significant threat to the patient’s life and contribute significantly to NF1 mortality [11].
PATHOGENESIS OF NEUROFIBROMAS
The Role of the Tumor Microenvironment in NF1-related Tumors
Currently, solid tumors are regarded increasingly often as complex organs, and researchers are paying more attention to the tumor environment, which is critical for tumor progression, metastasis, and drug resistance. Neurofibromin-deficient Schwann cells are identified as the primary neoplastic cells in NF1, while studies on murine models and patients have demonstrated that the loss of the NF1 gene in Schwann cells is insufficient for neurofibroma formation. Non-tumorigenic cells such as mast cells and hematopoietic effector cells, which are known to permeate in neurofibromas, also play an important role in tumor development and progression [13,14]. Interactions between tumorigenic cells and their surrounding microenvironment are critical for tumor progression as tumorigenic cells in neurofibromas hardly arise and grow in isolation.
Role of Ras Signaling in NF1-related Tumors
Neurofibromin acts as a Ras-GTPase activating protein that accelerates the intrinsic hydrolysis of Ras from active GTP-bound to inactive GDP-bound conformation. In NF1-related tumors, biallelic NF1 inactivity causes a complete loss of the functional activity of neurofibroma. With a complete loss of function of neurofibromin, Ras signaling sustains hyperactivity and results in the activity of diverse protein signaling networks, including Ras-MAPK and Ras/PI3K/AKT/mTOR pathway [15,16] (Fig. 1).
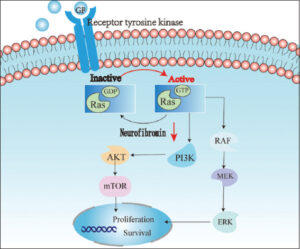 |
Figure 1: RAS signaling may be activated by the combination of growth factors and receptor tyrosine kinase. Neurofibromin accelerates the transformation of RAS from GTP-bound (active) to GDP-bound (inactive). In NF1-related tumors, the mutations of the NF1 gene result in an aberrant function of neurofibromin and increase the activity of Ras/Raf/MEK/ERK and Ras/PI3K/AKT/mTOR pathways, ultimately activating the RAS signaling pathways, which results in cell proliferation and survival. |
TREATMENT
At the early stage, clinical trials were aimed at mast cell function and angiogenesis, which were thought to be integral for the progression of neurofibromas in NF1. With further understanding of the pathogenesis of NF1-related tumors, more therapeutic trials attempted to focus on targeting the neoplastic Schwann cell and the tumor microenvironment [17].
In 2002, Packer et al. summarized the agents employed to treat NF1, including the antihistamine agent ketotifen fumarate, retinoic acid, interferon-alpha, thalidomide, and farnesyl protein transferase inhibitor [17]. According to that report, ketotifen fumarate and farnesyl protein transferase inhibitor are well tolerated. However, no patients experienced shrinkage of tumors, and the results were in accord with the subsequent clinical trials, which blocked their further application [18]. Later, a study on a NF1-deficient mouse model demonstrated that ketotifen fumarate indeed decreased mast cell infiltration, yet had no impact on mast cell numbers, degranulation, and tumor behavior [19]. The other three agents revealed symptomatic improvement and tumor shrinkage in several patients (Table 1). A phase 1 and 2 clinical trial of PEGylated interferon alpha-2b demonstrated partial responses (≥ 20% decrease from the baseline) in several patients, and time to progression (TTP) prolonged significantly to 29.4 months vs. 11.8 for the placebo group [20,21].
 |
Table 1: A summary and supplement of trials from Packer’s literature [17]. |
Those agents only showed low responses in several patients, which encouraged researchers to explore more effective agents. We will further discuss the efforts made in targeting the tumor environment and the signaling pathways in NF1-related tumors (Tables 2 and 3).
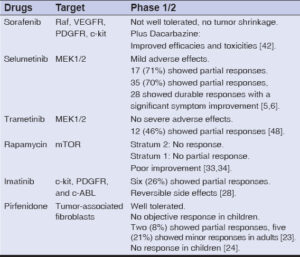 |
Table 2: Main results of clinical trials for NF1-related tumors. |
 |
Table 3: Clinical trials remaining to publish their outcomes. |
Efforts Targeting the Tumor Environment
Pirfenidone
Several studies implied that fibroblasts play an important role in the pathogenesis of NF1-related tumors [22]. A phase 2 clinical trial of pirfenidone, a broad-spectrum oral antifibrotic drug that modulates the expression of growth factors and cytokines relevant to fibrosis, was performed [23]. In this open-label pilot trial, pirfenidone at a total daily dose of 2400 mg proved to be effective in adult patients, which suggests that it deserves further investigation. However, in a phase 1/2 clinical trial of pirfenidone in children with NF1, neither tumor shrinkage nor significantly prolonged TTP were achieved, which was in contrast to the former trial [24].
Imatinib mesylate
Recent studies have revealed that a haploinsufficiency of NF1 and c-kit signaling in the hematopoietic system is required and sufficient for tumor progression [25]. Imatinib mesylate is a multitargeted c-kit, PDGFR, and c-ABL inhibitor and has been approved by the FDA for some tumors. It is the first medical treatment for PNFs targeting the stem cell factor/c-kit axis. A preclinical study demonstrated that imatinib mesylate not only reduced cell viability in vitro yet also inhibited cell proliferation and decreased tumor volume in xenograft models [26]. Yang et al. treated a cohort of adult Nf1flox/-;Krox20cre mice with imatinib, and the treatment group revealed regularly patterned Schwann cells, free of mast cell infiltrate, and reduced tumor volume [13].
Based on this efficacy, clinicians treated a three-year-old girl with 350 mg/m2 imatinib mesylate. After three months of treatment, a remarkable 70% reduction in tumor volume was observed, resolving the tumor-induced airway compression [13]. Khelifa et al. reported a case of a 42-year-old female with a 34-year history of NF1 and cutaneous vasculopathy that improved after treatment with imatinib [27].
Robertson et al. reported an open-label phase 2 trial of imatinib at 220 mg/m2 twice a day for pediatric patients and 400 mg for adults for at least six months in PNFs. In this trial, 23 patients completed the study and were evaluated. Six patients had a partial response, while the remaining thirteen patients withdrew prematurely. There was no significant difference between pediatric and adult patients [28].
The most common adverse effects included skin rash and edema. Other adverse effects such as reversible neutropenia and elevated aminotransferase were also noted in several patients. The patients in this study revealed poor compliance, which may be related to the biology of PNFs and the initial dosing of the drug [28].
Furthermore, a preclinical trial reported that nilotinib, a tyrosine kinase inhibitor, has several advantages and is more potent than imatinib in treating PNFs in vitro and in vivo [29].
Efforts in Targeting the Ras Signaling Pathway
The MAPK/ERK pathway has been one of the most important pathways for developing novel antitumor drugs. Initially, several drug discovery programs focused on farnesyltransferase inhibitors, which may prevent Ras locating to the membrane. Unfortunately, the disappointing clinical data prevented further investigation [18]. Currently, several compounds targeting the substrates of Ras are under clinical investigation and have made notable achievements.
Rapamycin
Rapamycin is an allosteric inhibitor of mTOR complex 1 and has been approved as an anti-rejection medication for transplantation [30]. Preclinical studies in patient-derived xenografts and genetically engineered mouse models demonstrated that rapamycin significantly inhibited the activity of mTOR and tumor growth [31,32].
Based on these findings, Weiss et al. conducted a 2-strata phase 2 clinical trial in NF1-associated non-progressive PNFs (stratum 2) and progressive PNFs (stratum 1), respectively [33,34]. In stratum 2, after six courses, no patients experienced disease improvement. However, the mean emotional and school domain scores revealed a significant increase [34]. The results differed from the preclinical trials. However, a lack of PN shrinkage was consistent with a preclinical study in which the administration of mTOR inhibitor, everolimus, did not cause a significant decrease in tumor volume [35]. This trial demonstrated that rapamycin could not cause tumor shrinkage in non-progressive PNFs, and further investigation of the efficacy on TTP was evaluated in stratum 1.
In stratum 1, after treatment, the subjects showed a partial response, with a maximum decrease of 17%. However, the estimated median TTP of the patients (15.4 months) was significantly longer than that of the placebo group (11.9 months) [33]. In addition, the study revealed some improvement in pain intensity. There were also cases reporting alleviated pain in patients with severe PN after receiving rapamycin [36].
Overall, the efficacy of rapamycin in reducing tumor volume was not especially satisfactory, yet considering the shortage of effective treatment options, rapamycin may be considered for selected patients. Further studies are required to identify subsets of PNFs that might be more likely to respond to rapamycin therapy and to explore combination therapies that may improve its efficacy.
Sorafenib
Sorafenib is a dual-action inhibitor that has been approved by the FDA for patients with advanced renal cell carcinoma or unresectable hepatocellular carcinoma [37]. Not only does it inhibit Raf kinase, yet also inhibits several receptor tyrosine kinases involved in neovascularization and proliferation, including VEGFR, PDGFR, and c-kit [38]. Sorafenib revealed a broad-spectrum antitumor activity in preclinical and clinical trials against numerous solid tumors. Similar results were obtained in clinical trials of progressive low-grade gliomas revealing high activation of MAPK pathways [39].
Little is known about NF1-related tumors, thus to explore the further application of sorafenib, Ambrosini et al. assumed that MPNSTs would be susceptible to this compound and conducted a preclinical trial with series of sarcoma cell lines. As a result, by suppressing the level of p-MEK and p-ERK, inhibiting cyclin D1 and pRb phosphorylation, sorafenib inhibited the growth of MPNST at low concentrations [40]. An in vivo study also illustrated that sorafenib significantly decreased MPNST volume by volumetric MRI with mild adverse reactions [35].
However, a phase 1 trial of the drug on children with PNFs demonstrated that, after an average of seven cycles, no tumor shrinkage was observed in nine patients enrolled in the study [41]. Considering the low single-agent response rates of sorafenib, D’Adamo et al. attempted sorafenib 400 mg oral twice daily plus dacarbazine 1000 mg/m2 intravenously, which achieved a modest clinical improvement with an eighteen-week disease control rate of 46%. However, combination therapy may increase the potential for significant toxicity [42].
Selumetinib
Initially, because of the lack of selective and potent Ras, Raf, and ERK inhibitors for most tumors, research on MEK inhibitors developed rapidly. Selumetinib is a second-generation MEK1/2 inhibitor. Mechanistically, with a non-competitive activity to ATP, MEK inhibitors bind to the binding site in MEK1/2, locking MEK1/2 into an inactive conformation, and thus prevent molecular interactions required for catalysis and ERK activation, consequently inhibiting cell proliferation and differentiation [43]. Selumetinib has shown promising potency and extensive antitumor activity in preclinical and clinical trials such as in glioma, gastrointestinal malignancies, thyroid cancers, NSCLCs, and melanomas. In preclinical models, MEK inhibitors have also been proven to be effective for PNFs. Walter et al. treated several genetically engineered mice with a highly specific MEK inhibitor PD0325901. At the end of the study, 80% of the mice experienced a striking reduction in neurofibroma volumes [44]. Then, Dombi et al. reported that selumetinib targeting MEK1/2 at a dose of 10 mg/kg twice daily on an intermittent dosing was also effective on a mouse model. At the end of the study, 12 out of 18 mice (67%) experienced a decrease in neurofibroma volume when compared to the baseline [5].
Based on these preclinical outcomes, Dombi et al. performed a phase 1 trial to test the clinical efficacy and safety of selumetinib in pediatric patients with inoperable, measurable plexiform neurofibromas [5]. In this trial, twenty-four children were treated with selumetinib at three dose levels—20 mg/m2, 25 mg/m2, and 30 mg/m2—twice daily on a continuous dosing schedule (cycles: 6–56). All patients experienced a decrease in tumor volume (average: 31%), and 17/24 (71%) patients had partial responses. Moreover, not only decreases in volume were achieved, yet also a significant symptomatic improvement was observed, such as in disfigurement, pain, and functional impairment. The most common side effects were mild, and some patients also experienced dose-limiting toxicities, including 4/12 patients in the 20 mg group, 3/6 patients in the 25 mg group, and 4/6 patients in the 30 mg group, yet all were resolvable [5].
Recently, a phase 2 trial of selumetinib for PNFs also showed a clinically meaningful improvement, with 35 patients (70%) having a confirmed partial response, among which 28 experienced a durable response (≥ 1 year). However, six patients suffered tumor progression and five patients discontinued the drug because of selumetinib-related toxicities [6]. A single-institution study confirmed that selumetinib was effective and safe for NF1-related PNFs, with all patients experiencing a sustained clinical improvement except one [45]. The exact efficacy in adult patients remains unclear, hence further investigation needs to be conducted.
Trametinib
Trametinib is a novel and highly specific allosteric MEK inhibitor. As monotherapy or combined therapy, it has been approved for numerous tumors harboring mutations in members of the MAPK pathway [46]. In 2019, Vaassen et al. reported an eleven-year-old girl suffering from a huge PNF that caused an extreme deformity, which benefited from trametinib therapy [47]. In that study, after six months of trametinib 0.5 mg twice daily, there was a 22% decrease in tumor volume, which made surgery possible.
McCowage et al. conducted a phase 1/2 single-arm multicohort trial that evaluated trametinib at 0.025–0.040 mg/kg/day in pediatric patients with unresectable NF1associated PNFs [48]. Twenty-six patients were recruited to the trial, and after a median duration of 61 weeks, twelve patients (46%) experienced a partial response, and no patients experienced severe adverse effects. The trial is still ongoing, yet reports have already demonstrated tolerability and clinical benefits in a cohort of children with plexiform neurofibromas.
Perreault et al. also presented a protocol for a phase 2 study investigating singleagent trametinib at a fixed dose of 0.025 mg/kg (≥ 6 years old) or 0.032 mg/kg (< 6 yrs. old) for eighteen cycles to evaluate the efficacy and safety in patients with pediatric low-grade gliomas and neurofibromas [49]. In this study, the authors expected to recruit a total of 150 patients, which included 46 patients with plexiform neurofibroma. At the end of the trial, they will evaluate not only standard response rates yet also progression-free survival, overall survival, and quality of life during treatment.
CONCLUSION
Despite neurofibromatosis-1 being a type of familial disease, a half of the reported cases are yet due to de novo mutation on chromosome primarily derived paternally, which may decrease quality of life and average life expectancy of the victim. Traditional therapeutic regimes for NF1-related tumors are limited to surgical removal or physical destruction, which by the involvement of adjacent tissues, particularly the nerve and vasculature, may complicate the procedure with ensuing frequently neoplasm recurrence that creates the urgency to explore new therapeutic methods of treating NF1-related tumors radically.
At the very beginning, experts focused on agents targeting the tumor microenvironment, such as pirfenidone and imatinib, yet there were low responses in patients with NF1. More efforts were made to explore small molecule compounds targeting RAS signaling pathways. By using these compounds, not only clinical symptoms were improved, yet also the shrinkage of tumors was achieved. Focusing on the development of precision oncology and the increased investigation of pathogenesis and molecular landscape of NF1-related tumors, more agents targeting MAPK/ERK and mTOR pathways are under investigation, with some having achieved inspiring outcomes.
As there is remarkable clinical efficacy of selumetinib in numerous other non-cutaneous tumors and some NF1-associated tumors, it may be considered for the tumor shrinkage of NF1 to not only shrink the volume, yet also improve other symptoms that may increase the quality of life of the patient. It has been proven to be effective against melanomas in some literature as well. Hence, more effort needs to be exerted to explore the specific mechanism of its action. We cannot ignore the fact that it may be considered for NF1 cases with large tumor sizes with an aim of, first, decreasing its size and volume then, subsequently, excising it. Furthermore, ERK1/2 inhibitors have recently produced inspiring results in preclinical research, especially in those tumors with acquired resistance to MEK inhibitors. Yet, they have been implicated less for NF1related tumors, hence more clinical research may be performed regarding its efficacy. Several systemic trials are currently being conducted to evaluate the efficacy of targeted therapeutics for cutaneous neurofibromas and many of them are still under investigation (Table 3). Only one phase 2 open-label, single-arm trial of everolimus (ClinicalTrials.gov; identifier: NCT02332902) revealed a significant reduction in lesion surface volume and PNF, yet no serious adverse events were accounted [50]. Hence, more clinical trials should be conducted with such agents to further study its outcomes with a life-time observation if possible.
REFERENCES
1. Anderson JL, Gutmann DH. Neurofibromatosis type 1. Handb Clin Neurol 2015;132:75-86.
2. Friedman JM. Neurofibromatosis 1:Clinical manifestations and diagnostic criteria. J Child Neurol. 2002;17:548-54;discussion 571-2, 646-51.
3. Lobbous M, Bernstock JD, Coffee E, Friedman GK, Metrock LK, Chagoya G, et al. An update on neurofibromatosis type 1-associated gliomas. Cancers (Basel). 2020;12:114.
4. Yunoue S, Tokuo H, Fukunaga K, Feng L, Ozawa T, Nishi T, et al. Neurofibromatosis type I tumor suppressor neurofibromin regulates neuronal differentiation via its GTPase-activating protein function toward Ras. J Biol Chem. 2003 Jul 18;278:26958-69.
5. Dombi E, Baldwin A, Marcus LJ, Fisher MJ, Weiss B, Kim A, et al. Activity of selumetinib in neurofibromatosis type 1-related plexiform neurofibromas. N Engl J Med. 2016;375:2550-60.
6. Gross AM, Wolters PL, Dombi E, Baldwin A, Whitcomb P, Fisher MJ, et al. Selumetinib in children with inoperable plexiform neurofibromas. N Engl J Med. 2020;382:1430-42.
7. Ortonne N, Wolkenstein P, Blakeley JO, Korf B, Plotkin SR, Riccardi VM, et al. Cutaneous neurofibromas:Current clinical and pathologic issues. Neurology. 2018;91(2 Suppl 1):S5-S13.
8. Jouhilahti EM, Peltonen S, Callens T, Jokinen E, Heape AM, Messiaen L, et al. The development of cutaneous neurofibromas. Am J Pathol. 2011;178:500-5.
9. Granström S, Langenbruch A, Augustin M, Mautner VF. Psychological burden in adult neurofibromatosis type 1 patients:Impact of disease visibility on body image. Dermatology. 2012;224:160-7.
10. Korf BR. Plexiform neurofibromas. Am J Med Genet. 1999;89:31-7.
11. Evans DG, Baser ME, McGaughran J, Sharif S, Howard E, Moran A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J Med Genet. 2002;39:311-4.
12. Ferner RE, Gutmann DH. International consensus statement on malignant peripheral nerve sheath tumors in neurofibromatosis. Cancer Res. 2002;62:1573-7.
13. Yang FC, Ingram DA, Chen S, Zhu Y, Yuan J, Li X, et al. Nf1-dependent tumors require a microenvironment containing Nf1+/– and c-kit-dependent bone marrow. Cell. 2008;135:437-48.
14. Zhu Y, Ghosh P, Charnay P, Burns DK, Parada LF. Neurofibromas in NF1:Schwann cell origin and role of tumor environment. Science. 2002;296:920-2.
15. Brems H, Beert E, de Ravel T, Legius E. Mechanisms in the pathogenesis of malignant tumours in neurofibromatosis type 1. Lancet Oncol. 2009;10:508-15.
16. Dasgupta B, Yi Y, Chen DY, Weber JD, Gutmann DH. Proteomic analysis reveals hyperactivation of the mammalian target of rapamycin pathway in neurofibromatosis 1-associated human and mouse brain tumors. Cancer Res. 2005;65:2755-60.
17. Packer RJ, Gutmann DH, Rubenstein A, Viskochil D, Zimmerman RA, Vezina G, et al. Plexiform neurofibromas in NF1:Toward biologic-based therapy. Neurology. 2002;58:1461-70.
18. Widemann BC, Dombi E, Gillespie A, Wolters PL, Belasco J, Goldman S, et al. Phase 2 randomized, flexible crossover, double-blinded, placebo-controlled trial of the farnesyltransferase inhibitor tipifarnib in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Neuro Oncol. 2014;16:707-18.
19. Burks CA, Rhodes SD, Bessler WK, Chen S, Smith A, Gehlhausen JR, et al. Ketotifen modulates mast cell chemotaxis to kit-ligand, but does not impact mast cell numbers, degranulation, or tumor behavior in neurofibromas of NF1-deficient mice. Mol Cancer Ther. 2019;18:2321-30.
20. Jakacki RI, Dombi E, Potter DM, Goldman S, Allen JC, Pollack IF, et al. Phase I trial of pegylated interferon-alpha-2b in young patients with plexiform neurofibromas. Neurology. 2011;76:265-72.
21. Jakacki RI, Dombi E, Steinberg SM, Goldman S, Kieran MW, Ullrich NJ, et al. Phase II trial of pegylated interferon alfa-2b in young patients with neurofibromatosis type 1 and unresectable plexiform neurofibromas. Neuro Oncol. 2017;19:289-97.
22. Sasaki T, Arai K, Nagai Y. Growth and collagen synthesis of cultured neurofibroma fibroblasts. J Dermatol. 1992;19:598-601.
23. Babovic-Vuksanovic D, Ballman K, Michels V, McGrann P, Lindor N, King B, et al. Phase II trial of pirfenidone in adults with neurofibromatosis type 1. Neurology. 2006;67:1860-2.
24. Widemann BC, Babovic-Vuksanovic D, Dombi E, Wolters PL, Goldman S, Martin S, et al. Phase II trial of pirfenidone in children and young adults with neurofibromatosis type 1 and progressive plexiform neurofibromas. Pediatr Blood Cancer. 2014;61:1598-602.
25. Staser K, Yang FC, Clapp DW. Mast cells and the neurofibroma microenvironment. Blood. 2010;116:157-64.
26. Demestre M, Herzberg J, Holtkamp N, Hagel C, Reuss D, Friedrich RE, et al. Imatinib mesylate (Glivec) inhibits Schwann cell viability and reduces the size of human plexiform neurofibroma in a xenograft model. J Neurooncol. 2010;98:11-9.
27. Khelifa I, Saurat JH, Prins C. Use of imatinib in a patient with cutaneous vasculopathy in the context of von Recklinghausen disease/neurofibromatosis. Br J Dermatol. 2015;172:253-6.
28. Robertson KA, Nalepa G, Yang FC, Bowers DC, Ho CY, Hutchins GD, et al. Imatinib mesylate for plexiform neurofibromas in patients with neurofibromatosis type 1:A phase 2 trial. Lancet Oncol. 2012;13:1218-24.
29. Wei J, Freytag M, Schober Y, Nockher WA, Mautner VF, Friedrich RE, et al. Nilotinib is more potent than imatinib for treating plexiform neurofibroma in vitro and in vivo. PLoS One. 2014;9:e107760.
30. Saunders RN, Metcalfe MS, Nicholson ML. Rapamycin in transplantation:A review of the evidence. Kidney Int. 2001;59:3-16.
31. Bhola P, Banerjee S, Mukherjee J, Balasubramanium A, Arun V, Karim Z, et al. Preclinical in vivo evaluation of rapamycin in human malignant peripheral nerve sheath explant xenograft. Int J Cancer. 2010;126:563-71.
32. Johansson G, Mahller YY, Collins MH, Kim MO, Nobukuni T, Perentesis J, et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol Cancer Ther. 2008;7:1237-45.
33. Weiss B, Widemann BC, Wolters P, Dombi E, Vinks A, Cantor A, et al. Sirolimus for progressive neurofibromatosis type 1-associated plexiform neurofibromas:A neurofibromatosis Clinical Trials Consortium phase II study. Neuro Oncol. 2015;17:596-603.
34. Weiss B, Widemann BC, Wolters P, Dombi E, Vinks AA, Cantor A, et al. Sirolimus for non-progressive NF1-associated plexiform neurofibromas:An NF clinical trials consortium phase II study. Pediatr Blood Cancer. 2014;61:982-6.
35. Wu J, Dombi E, Jousma E, Scott Dunn R, Lindquist D, Schnell BM, et al. Preclincial testing of sorafenib and RAD001 in the Nf(flox/flox);DhhCre mouse model of plexiform neurofibroma using magnetic resonance imaging. Pediatr Blood Cancer. 2012;58:173-80.
36. Hua C, Zehou O, Ducassou S, Minard-Colin V, Hamel-Teillac D, Wolkenstein P, et al. Sirolimus improves pain in NF1 patients with severe plexiform neurofibromas. Pediatrics. 2014;133:e1792-7.
37. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Siebels M, et al. Sorafenib in advanced clear-cell renal-cell carcinoma. N Engl J Med. 2007;356:125-34.
38. Wilhelm SM, Carter C, Tang L, Wilkie D, McNabola A, Rong H, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004;64:7099-109.
39. Karajannis MA, Legault G, Fisher MJ, Milla SS, Cohen KJ, Wisoff JH, et al. Phase II study of sorafenib in children with recurrent or progressive low-grade astrocytomas. Neuro Oncol. 2014;16:1408-16.
40. Ambrosini G, Cheema HS, Seelman S, Teed A, Sambol EB, Singer S, et al. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol Cancer Ther. 2008;7:890-6.
41. Kim A, Dombi E, Tepas K, Fox E, Martin S, Wolters P, et al. Phase I trial and pharmacokinetic study of sorafenib in children with neurofibromatosis type I and plexiform neurofibromas. Pediatr Blood Cancer. 2013;60:396-401.
42. D’Adamo DR, Dickson MA, Keohan ML, Carvajal RD, Hensley ML, Hirst CM, et al. A phase II trial of sorafenib and dacarbazine for leiomyosarcoma, synovial sarcoma, and malignant peripheral nerve sheath tumors. Oncologist. 2019;24:857-63.
43. Yeh TC, Marsh V, Bernat BA, Ballard J, Colwell H, Evans RJ, et al. Biological characterization of ARRY-142886 (AZD6244), a potent, highly selective mitogen-activated protein kinase kinase 1/2 inhibitor. Clin Cancer Res. 2007;13:1576-83.
44. Jessen WJ, Miller SJ, Jousma E, Wu J, Rizvi TA, Brundage ME, et al. MEK inhibition exhibits efficacy in human and mouse neurofibromatosis tumors. J Clin Invest. 2013;123:340-7.
45. Espírito Santo V, Passos J, Nzwalo H, Carvalho I, Santos F, Martins C, et al. Selumetinib for plexiform neurofibromas in neurofibromatosis type 1:A single-institution experience. J Neurooncol. 2020;147:459-63.
46. Kondyli M, Larouche V, Saint-Martin C, Ellezam B, Pouliot L, Sinnett D, et al. Trametinib for progressive pediatric low-grade gliomas. J Neurooncol. 2018;140:435-44.
47. Vaassen P, Dürr N, Röhrig A, Willing R, Rosenbaum T. Trametinib induces neurofibroma shrinkage and enables surgery. Neuropediatrics. 2019;50:300-3.
48. McCowage GB, Mueller S, Pratilas CA, et al. Trametinib in pediatric patients with neurofibromatosis type 1 (NF-1)-associated plexiform neurofibroma:A phase I/IIa study. J Clin Oncol. 2018;36.
49. Perreault S, Larouche V, Tabori U, Hawkin C, LippéS, Ellezam B, et al. A phase 2 study of trametinib for patients with pediatric glioma or plexiform neurofibroma with refractory tumor and activation of the MAPK/ERK pathway:TRAM-01. BMC Cancer. 2019;19:1250.
50. Slopis JM, Arevalo O, Bell CS, Hebert AA, Northrup H, Riascos RF, et al. Treatment of disfiguring cutaneous lesions in neurofibromatosis-1 with everolimus:A phase II, open-label, single-arm trial. Drugs R D. 2018;18:295-302.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
|

Comments are closed.