Spinulosis revealing dermatomyositis relapse: A new description of Wong’s dermatomyositis
Mehdi Khallaayoune , Fatima Azzahra Elgaitibi, Siham Belmourida, Mariame Meziane, Nadia Ismaili, Leila Benzekri, Karima Senouci
, Fatima Azzahra Elgaitibi, Siham Belmourida, Mariame Meziane, Nadia Ismaili, Leila Benzekri, Karima Senouci
Department of Dermatology, Ibn Sina University Hospital, Mohammed V University, Rabat, Morocco
Corresponding author: Mehdi Khallaayoune, MD
How to cite this article: Khallaayoune M, Elgaitibi FA, Belmourida S, Meziane M, Ismaili N, Benzekri L, Senouci K. Spinulosis revealing dermatomyositis relapse: A new description of Wong’s dermatomyositis. Our Dermatol Online. 2022;13(2):227-228.
Submission: 04.02.2021; Acceptance: 12.05.2021
DOI: 10.7241/ourd.20222.31
Citation tools:

Copyright information
© Our Dermatology Online 2022. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
Sir,
Dermatomyositis (DM) is a systemic autoimmune disease affecting characteristically the skin and muscles. The typical cutaneous features include Gottron’s papules, a heliotrope rash, and edema of the eyelids. Spinulosis is an unusual sign, not raising suspicion for a DM diagnosis. Herein, we report a case of DM relapsing as Wong’s dermatomyositis (WDM) as an isolated spinulosic eruption.
A 42-year-old female presented with a pruritic papular eruption involving both arms and the back persistent for the last two months. An anamnesis included a history of DM diagnosed five years earlier, in complete remission under the administration of corticosteroids for three years. A physical examination revealed erythematous and violaceous scaly plaques resulting from confluent follicular papules, resembling pityriasis rubra pilaris (PRP) on the lateral sides of the arms (Fig. 1) and the upper back (Fig. 2). Muscular testing was subnormal and the general status was conserved. Laboratory findings revealed a high level of blood CPK (3×N), LDH (4×N), and ASAT (3.5×N), while electromyography displayed myogenic changes. A muscular biopsy revealed necrotic muscular cells with perifascicular atrophy and lymphocytic inflammatory infiltrate. A histological skin examination found follicular hyperkeratosis with perifollicular interface dermatitis and interstitial mucin deposits in the dermis (Fig. 3). Based on these findings, the diagnosis of WDM was retained. A malignancy workup was negative. Corticosteroid therapy was initiated with prednisone 1 mg/kg/day in combination with hydroxychloroquine 200 mg twice a day, resulting in a remarkable clinical and biological improvement.
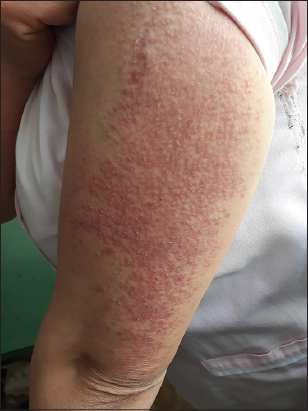 |
Figure 1: Confluent spinulosic scaly papules resulting in the plaque on the arm. |
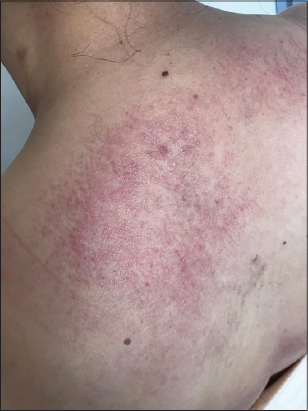 |
Figure 2: Violaceous plaque with follicular papules on the upper back. |
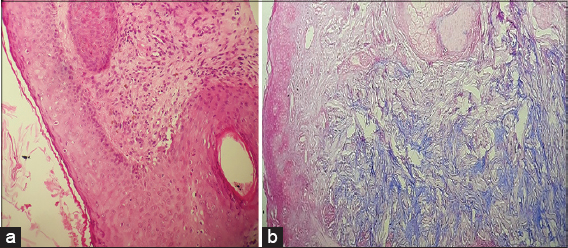 |
Figure 3: (a) Perifollicular interface dermatitis (b) with dermal mucin deposits (Alcian blue staining). |
WDM is characterized by the presence of PRP-like follicular papules in a patient with dermatomyositis (DM) [1]. Some authors suggest that this exceptional presentation should be considered a true association of DM with PRP rather than a variant of DM [2]. In this rare form, spinulosic lesions are mainly located on the trunk or limbs with occasional palmoplantar keratoderma [2]. Follicular lesions are usually associated with the typical signs of DM but some cases of isolated PRP features related to WDM have been reported [3]. To our knowledge, none has been reported as a relapse of a previous classic DM. Our patient relapsed showing spinulosic lesions absent in the prior episode five years earlier, not displaying at this time any classic cutaneous signs of DM. Such puzzling presentations are very unusual and may be confusing for physicians. In our patient, the diagnosis was retained on the basis of the histopathological findings of interface dermatitis and interstitial mucin deposits in the dermis, increased serum levels of muscle enzymes with a myogenic electromyographic pattern, the typical muscular histological changes, and a medical history of DM diagnosed five years earlier. In WDM, histology usually reveals follicular hyperkeratosis with keratotic plugs filling dilated follicular infundibula, which may raise several differential diagnoses, including PRP, discoid lupus erythematosus (DLE), and, to a lesser degree, follicular psoriasis and keratosis pilaris [4]. Interface dermatitis and mucin deposits, as in our patient, are the hallmarks of distinguishing DM from PRP, follicular psoriasis, and keratosis pilaris. Although these findings may also be observed in DLE [4], in our patient, there were no clinical or histological arguments supporting DLE rather than a diagnosis of DM. Myositis as, well as interstitial lung disease, appears to be comparable to the classic variant [5]. The risk of associated internal malignancy is unknown but could be lower than in the classic form [4]. Treatment and management do not differ from classic dermatomyositis.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Didona D, Fania L, Didona B. Wong-type dermatomyositis. G Ital Dermatol Venereol. 2018;153:115-6.
2. Haro R, Revelles JM, Fariña Mdel C, Martín L, Requena L. Wong’s dermatomyositis:A new case and review of the literature. Int J Dermatol. 2013;52:466-70.
3. Umanoff N, Fisher A, Carlson JA. Wong-type dermatomyositis showing porokeratosis-like changes (columnar dyskeratosis):A case report and review of the literature. Dermatopathology (Basel). 2015;2:1-8.
4. Mutasim DF, Egesi A, Spicknall KE. Wong-type dermatomyositis:A mimic of many dermatoses. J Cutan Pathol. 2016;43:781-6.
5. Canavan T, Sidorsky T, Doan LT, Ricardo-Gonzalez RR, Shen G, Rosenblum MD. A case of Wong-type dermatomyositis with concomitant anti-MDA5 features. J Am Acad Dermatol. 2014;70:62-4.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-0313-6152 http://orcid.org/0000-0002-2795-9161 http://orcid.org/0000-0002-0313-6152 http://orcid.org/0000-0002-2795-9161 |

Comments are closed.