Papular mucinosis (papular lichen myxedematosus): Clinical and histopathological evaluation
Khalifa Sharquie 1, Raed I Jabbar2
1, Raed I Jabbar2
1Department of Dermatology, College of Medicine, University of Baghdad. Medical City Teaching Hospital, Baghdad, Iraq, 2Department of Dermatology, Falluja Teaching Hospital, Al-Anbar Health Directorate, Anbar, Iraq
Corresponding author: Prof. Khalifa Sharquie, MD, PhD, FRCP Edin
Submission: 06.08.2020; Acceptance: 11.10.2020
DOI: 10.7241/ourd.20212.3
Cite this article: Sharquie K, Jabbar RI. Papular mucinosis (papular lichen myxedematosus): Clinical and histopathological evaluation. Our Dermatol Online. 2021;12(2):113-119.
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Background: Papular mucinosis is a variety of mucinosis characterized by excessive production of mucin by fibroblasts and deposition in the dermis. It manifests itself with fleshy papules or plaques in different sites of the body and taking different clinical morphological cutaneous features.
Objective: The objective is to report and evaluate the different clinical and histopathological features of the disease in Iraqi patients.
Patients and Methods: This is a case series and a clinical descriptive study in which ten patients with papular mucinosis were reported during the period from 2012 through 2019. The age ranged from 4 to 56 years, with seven females and three males. Clinical evaluation regarding histories of the disease and examination was carried out. General investigation was done and skin biopsy for histopathological assessment was conducted.
Results: Nine patients were adults, with their age ranging from 20 to 56 years, a mean of around 35 years, and only one 4-year-old child. It is a disease with a female predominance, as observed in 7 (70%) females. The common sites of involvement were the face but the rash may extend to affect the neck and upper arms. The rash appeared in the form of skin-colored or red fleshy papules and plaques or in diffuse erythematous orange peel-like forms. The rash was asymptomatic in most patients. The pathology of the disease clarified the diffuse deposition of mucin in the dermis, as was demonstrated with H&E staining.
Conclusion: Papular mucinosis is a rare disease characterized by mucin deposition in the skin affecting mostly adult females. The face is commonly involved together with other areas such as the neck and upper arms, but the trunk and lower limbs are spared. It manifests itself with different clinical morphological cutaneous features. Papular granuloma annulare must be considered as an important differential diagnosis in all cases of papular mucinosis.
Key words: Lichen myxedematosus; Papular mucinosis; Mucin
INTRODUCTION
The cutaneous mucinoses are a heterogeneous cluster of disorders in which an atypical amount of mucin conglomerates in the skin, either diffusely or focally [1].
Mucin is an element of the dermal extracellular matrix and is usually manufactured in little quantities by fibroblasts. It is a jelly-like, amorphous compound of acid glycosaminoglycans. The acid glycosaminoglycans may be connected to both sides of the protein core, as in the case of dermatan sulfate and chondroitin sulfate, or they may be free, as in the case of hyaluronic acid, which is the most important constituent of dermal mucin. Mucin is capable of holding water in quantities of up to 1000 times its weight, playing an important role in maintaining the water and salt equilibrium of the dermis. In routinely stained sections, the existence of either a blue-staining material between separated collagen bundles or empty spaces within the dermis are important hints of mucin deposition. For verification, special stains can be utilized, such as Alcian blue, toluidine blue, and colloidal iron [2].
Pathogenesis
The exact pathogenesis of mucinosis is unknown. The most agreed hypothesis is that circulating cytokines, such as tumor necrosis factor alpha, transforming growth factor beta, and Interleukin-1, intensify the reproduction of fibroblasts and glycosaminoglycan production. In vitro, the serum of patients with scleromyxedema can enhance the production of DNA by fibroblasts [3].
Classification
Cutaneous mucinoses may be classified as primary, in which mucin deposition is the major histological feature that leads to clinically unique lesions, and secondary, in which mucin constitutes an additional associated histological finding. Primary mucinoses are then subdivided into degenerative–inflammatory forms and hamartomatous–neoplastic forms. The degenerative–inflammatory forms are further subdivided into dermal and follicular [4].
Classification of Primary Mucinoses
A-Degenerative–Inflammatory Mucinoses:
1. Dermal
a. Scleromyxedema
b. Localized variants of lichen myxedematosus (LM):
- Discrete papular mucinosis
- Acral persistent papular mucinosis
- Cutaneous mucinosis of infancy (CMI)
- Nodular mucinosis
c. Self-healing cutaneous mucinosis
- Juvenile type
- Adult type
d. Scleredema
- Nondiabetic (types I and II)
- Diabetic (type III)
e. Mucinoses associated with altered thyroid function
- Localized (pretibial) myxedema
- Generalized myxedema
f. Reticular erythematous mucinosis
g. Papulonodular mucinosis associated with autoimmune connective tissue disease
h. Digital mucous cyst (myxoid cyst)
i. Cutaneous focal mucinosis
j. Miscellaneous mucinoses
2. Follicular
- Follicular mucinosis
- Urticaria-like follicular mucinosis
B. Hamartomatous–Neoplastic Mucinoses:
- (Angio)myxoma
- Mucinous nevus
Classification of Secondary Mucinoses (Associated with Histological Deposition of Mucin)
1. Epithelial mucinosis
- Basal cell carcinoma
- Rare: Squamous cell carcinoma, verruca, seborrheic keratosis, keratoacanthoma, mycosis fungoides.
2. Dermal mucinosis
- Granuloma annulare
- Lupus erythematosus, scleroderma and dermatomyositis
- Epithelial tumors (e.g., basal cell carcinoma, eccrine carcinoma)
- Mesenchymal tumors (e.g., myxosarcoma, myxoid lipoblastoma)
- Neural tumors (e.g., neurofibroma, lobular neuromyxoma)
- Other tumors (e.g., cutaneous metastases, mucinous carcinoma of the eyelid)
- Hypertrophic scarring
- Obesity-associated lymphedema
- Rare: chronic graft-versus-host disease, cutaneous reactions to interferon, herpes zoster, venous insufficiency.
3. Follicular mucinosis
- Eczematous dermatoses
- Mycosis fungoides
- Rare: lupus erythematosus, insect bites, familial reticuloendotheliosis, side effect of imatinib.
Scleromyxedema
Scleromyxedema is a rare cutaneous disorder characterized by dispersed eruption of hard, waxy, dome-shaped or flat-topped papules and nodules 2 – 3 mm in size that may amalgamate to form plaques and that involve the head, neck, trunk, and extremities. The scalp and mucosa are usually uninvolved [5]. Papules are generally arranged linearly and the neighboring skin exhibits a sclerodermoid manifestation. There is no significant gender predominance: males and females are equally affected. It most commonly occurs in adults between the fifth and sixth decades of life [6]. Mucin deposition in the dermis is responsible for the skin features. A sclerodermoid eruption with multiple papules, edema, and erythema—as well as papular induration over the ears and glabella—confer a leonine facies [7,8]. Skin thickening in chronic scleromyxedema can lead to decreased articular motion and difficulty in opening the mouth. Itching and dysesthesia are usual complaints as well [6,7].
Patients with scleromyxedema may have a number of systemic manifestations, such as muscular, rheumatologic, neurologic, renal, pulmonary, and cardiovascular [7].
Diagnostic criteria for scleromyxedema are as follows: a) histological triad of mucin deposition, fibroblast reproduction, and fibrosis; b) monoclonal gammopathy, predominantly IgG l and less commonly IgG k; c) absence of thyroid diseases [4].
Pathology
Scleromyxedema is characterized by a triad of pathologic features: [9]
- a diffuse accumulation of mucin in the upper and mid reticular dermis;
- an increase in collagen deposition;
- a noticeable proliferation of irregularly arranged fibroblasts.
Localized Variants of Lichen Myxedematosus (LM)
In the localized variants of LM, patients show small, firm, waxy papules (or nodules and plaques created by coalescence of the papules) that involve several sites, usually the upper and lower limbs and/or the trunk. The skin is the only site of involvement and these variants, in contrast to scleromyxedema, are not accompanied by sclerosis, paraproteinemia, or systemic involvement, nor are they accompanied by a thyroid disorder [10].
The diagnostic criteria of the localized variants of LM include a papular or nodular eruption and the histopathological triad (mucin accumulation, fibroblast reproduction at different grades) without monoclonal gammopathy or thyroid diseases [4].
Epidemiology
The exact incidence and prevalence rates of the localized variants of lichen myxedematosus remain unknown [4].
CLINICAL FEATURES
Discrete Papular Lichen Myxedematosus (DPLM)
DPLM is characterized by 2 – 5 mm papules, numbering from several to hundreds and affecting the extremities and trunk in a symmetrical fashion [12]. The involved skin is not indurated and the face usually uninvolved. Progression occurs slowly and without systemic involvement. Although prognosis is good, spontaneous resolution is rare [12,13].
Acral Persistent Papular Mucinosis (APPM)
In APPM, multiple ivory to skin-colored papules involve exclusively the dorsal aspects of the hands and extensor surfaces of the distal forearms and show a female predominance (female : male = 3 : 1). Lesions are persistent but without systemic involvement [14].
Cutaneous Mucinosis of Infancy (CMI)
In CMI, hard, opalescent papules involve the neck, upper arms (especially the elbows), and trunk [10]. In contrast to self-healing mucinosis, neither systemic manifestations nor spontaneous remission are observed. Of the handful of cases described to date, at least two have had a congenital linear variant, which might be better classified as an example of a mucinous nevus [15].
Nodular Lichen Myxedematosus
Nodular LM is characterized by firm, skin-colored nodules usually more than 6 mm in diameter with or without papules located on the trunk and extremities. Very few cases of nodular LM have been reported and the age of onset seems to be lower compared to APPM and DPLM [16–18].
Atypical Forms of Lichen Myxedematosus
Occasionally, patients with LM have abnormal manifestations or manifestations intermediate between scleromyxedema and localized LM [10].
Pathology
In the localized forms of LM, the histological changes are less distinctive than in scleromyxedema. Mucin accumulates in the upper and mid reticular dermis and fibroblast reproduction is variable; fibrosis is not noticeable and may even not seen. In acral persistent papular mucinosis, mucin accumulates focally in the upper part of the dermis with no increase in the number of fibroblasts [9].
Differential Diagnosis
Pathological examination of the skin assists in differentiating the localized types of LM from several papular lesions that can have the same appearance, such as papular granuloma annulare, colloid milium, papular elastorrhexis, molluscum contagiosum, amyloidosis, and eruptive collagenomas [4].
Treatment
Localized LM does not require therapy, and a wait-and-see approach is recommended. Topical therapy with corticosteroids or calcineurin inhibitors may be of some benefit [11]. One patient with associated human immunodeficiency virus (HIV) infection had a complete remission after treatment with oral isotretinoin. Spontaneous remission may occur [19], even with an HIV infection [20].
Complete resolution of LM nodules has been reported with intralesional injection of triamcinolone acetonide at a dose of 0.02 – 0.05 mL (8 mg/mL) [18].
Destructive modalities, including carbon dioxide (CO2) laser, erbium-doped yttrium aluminum garnet (Er-YAG) laser, cryotherapy, electrocoagulation, and electrofulguration, are acceptable cosmetic treatments for the localized types [21].
Self-Healing Cutaneous Mucinosis
Self-healing cutaneous mucinosis was initially reported in children, with the age ranging from 1 to 15 years [2]. The same disorder, thereafter, appeared in several adults [22].
Clinical manifestations are as follows: a) an acute eruption of several papules, sometimes collecting into linear infiltrated plaques, on the face, neck, scalp, abdomen, and thighs; and b) mucinous subcutaneous nodules in the periarticular areas and the face with periorbital swelling. In addition, systemic symptoms (e.g., fever, muscle tenderness, and arthralgias) may occur with skin lesions, but paraproteinemia, bone marrow plasmacytosis, and thyroid disorders are absent. Spontaneous remission over a period of 1 – 8 months is characteristic. Histologically, papular lesions show mucin deposition in the dermis with mild inflammation and some increase in the number of fibroblasts, whereas nodules have deep mucinous deposits accompanied by bands of fibrosis and obvious reproduction of fibroblast-like cells and gangliocyte-like cells resembling proliferative fasciitis [23].
Patients and Methods
This is a case series and a clinical descriptive study in which ten patients with papular mucinosis were collected at the Dermatology Center in Baghdad Medical City, Baghdad, Iraq, during the period from July 2012 through December 2019.
The age ranged from 4 to 56 years. The patients were seven females and three males.
Formal approval was taken from each patient after the nature, course, prognosis, and aim of the study were explained to each of the patients prior to the commencement of the study.
A full history was taken from each patient, including sex, age, age of disease onset, duration of the disease, associated skin and systemic symptoms, and past medical and drug history.
Proper clinical examination was conducted to determine the sites, colors, sizes, clinical appearances, and morphological distributions of the lesions.
A biopsy was taken from five patients from selected sites and processed for histopathology with H&E staining.
Pathological and clinical pictures were studied together for clinical and pathological correlation.
Erythrocyte sedimentation rate, complete blood count, blood urea, and serum creatinine were measured for each patient.
RESULTS
Clinical Results
Nine patients (90%) were adults, with the age ranging from 20 to 56 years, a mean of around 35 years, and only one 4-year-old child. The disease shows a female predominance, as observed in 7 (70%) females. The common sites of involvement were the face but the rash may extend to affect the neck and upper arms, but not the trunk and lower limbs. Lesions appeared in the form of fleshy, skin-colored or red papules and plaques or in diffuse erythematous orange peel-like forms (Figs. 1 and 2).
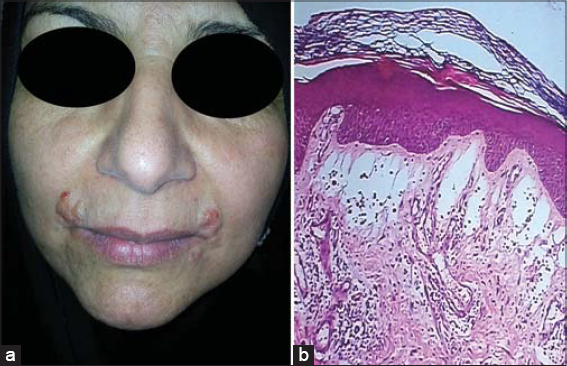 |
Figure 1: A 46-year-old female with (a) plaques of localized lichen myxedematosus around the mouth angles and (b) an H&E-stained section from the same patient showing mucin deposition in the papillary and reticular dermis together with hyperkeratosis and acanthosis of the epidermis (H&E, 10×). |
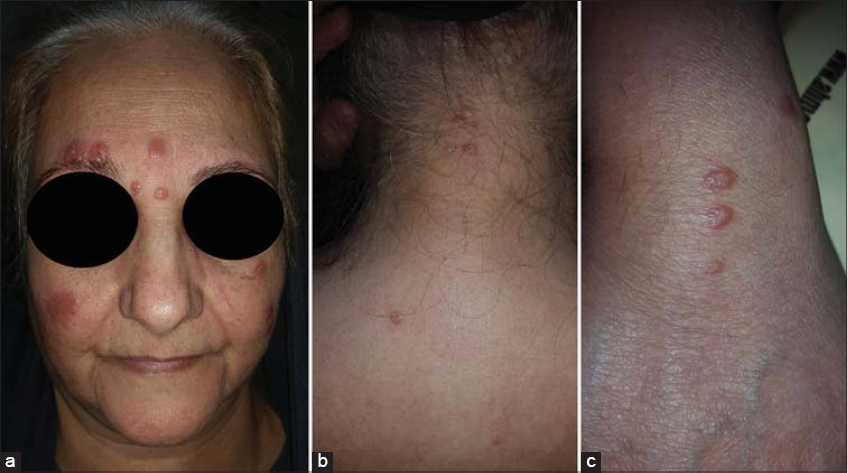 |
Figure 2: A 56-year-old female with even papulonodular plaques of localized lichen myxedematosus affecting (a) the face, (b) the posterior neck and upper trunk, and (c) the forearm. |
The rash was asymptomatic in most patients and only 2 (20%) experienced mild itching.
A 4-year-old female showed an acute eruption of asymptomatic papulonodular skin lesions persistent for two months and involving the face, scalp, neck, trunk, and extremities with swelling under the eyes. Some lesions had healed spontaneously during the course of the disease. A skin biopsy revealed mucin deposition in the superficial dermis. Clinical features and pathological findings of this case were matched together and diagnosed as self-healing juvenile cutaneous mucinosis (Fig. 3).
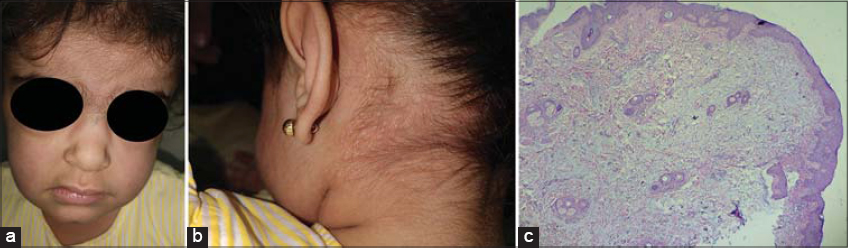 |
Figure 3: Self-healing juvenile cutaneous mucinosis in a four-year-old female involving (a) the face and forehead and (b) the postauricular and posterior neck. (c) An H&E-stained section showing diffuse mucin deposition in the papillary and reticular dermis with thinning of the epidermis (H&E, 10×). |
No systemic symptoms appeared in any patient except mild fever in case of self-healing juvenile cutaneous mucinosis. All results from laboratory investigations were normal.
Topical steroids were used with some beneficial results. In one case with a facial rash, diathermy was applied with favorable cosmetic results.
Pathological Results
Biopsies of the skin that stained with H&E clarified the diffuse deposition of mucin in the superficial and deep dermis and in direct contact with dermoepidermal junction. However, there was no increase in the fibroblast cells and no collagen deposition was noticed. The changes in the epidermis were variable, as acanthosis and hyperkeratosis were observed in some cases, while thinning of the epidermis in others (Figs. 1 and 3).
DISCUSSION
To the best of our knowledge, this is the first study on localized lichen myxedematosus (papular mucinosis) showing ten cases with different variants of the condition, with previous studies being either case reports [5,11–13,18] or a study with one type of localized cutaneous mucinosis [24].
Lichen myxedematosus is a rare cutaneous disorder as confirmed by the present work: ten patients have been seen by one doctor during a period of eight years.
In this study, 9 (90%) patients were adults, with the age ranging from 20 to 56 years. The condition studied has a female predominance as 70% of our cases were females. These results cannot be compared to other studies because of the rarity of studies on localized lichen myxedematosus and because case reports of only one or two varieties have been recorded. The exact incidence and preponderance of the different varieties of localized lichen myxedematosus are unknown because of their rarity. A female dominance has only been reported in acral persistent papular mucinosis, which is comparable to our findings [4].
In our work, the common sites of involvement were the face, but the rash may extend to affect the neck and upper arms. With the exception of a case of self-healing juvenile cutaneous mucinosis, the morphological distribution of lesions in most of our cases did not match any variant of localized lichen myxedematosus. This variability in morphological distribution cannot be explained but might have had a racial element or might have been a new entity of localized cutaneous mucinosis.
Histopathological results showed diffuse deposition of mucin in the papillary and reticular dermis and encroaching the dermoepidermal junction, and these results are comparable with what has been reported, but fibroblast cells proliferation and collagen deposition have not been observed in our work [9,11–15].
Regarding the differential diagnosis of papular lichen myxedematosus, papular granuloma annulare must be excluded [4].
Regarding the treatment of localized lichen myxedematosus, topical steroids, topical calcineurin inhibitors, and intralesional steroids have been used in previous reports. In our study, topical steroids were used with some beneficial results. In one case with a facial rash, diathermy was applied with favorable cosmetic results. Regarding the case of self-healing juvenile cutaneous mucinosis, no treatment was needed, except for reassurance of the parents as it is a self-limiting disease.
CONCLUSION
Papular lichen myxedematosus mucinosis is a rare disease characterized by mucin deposition in the skin, affecting mostly adult females. The face is commonly involved together with other sites such as the neck and upper arms. The condition presents itself with different clinical morphological cutaneous features. Granuloma annulare of the papular variety must be excluded in all cases of papular mucinosis.
Statement of Human and Animal Rights
All the procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the 2008 revision of the Declaration of Helsinki of 1975.
Statement of Informed Consent
Informed consent for participation in this study was obtained from all patients.
REFERENCES
1. Rongioletti F. Mucinoses. In:Rongioletti F, Smoller BR, editors. Clinical and Pathological Aspects of Skin Diseases in Endocrine, Metabolic, Nutritional and Deposition Disease. New York:Springer. 2010;146-7.
2. Warner TF, Wrone DA, Williams EC, Cripps DJ, Mundhenke C, Friedl A. Heparan sulphate proteoglycan in scleromyxedema promotes FGF-2 activity. Pathol Res Pract. 2002;198:701-7.
3. Rongioletti F, Rebora A. Mucinoses. In:Bolognia JL, Jorizzo JJ, Schaffer JV. Dermatology. 3rd ed. Philadelphia:Elsevier Saunders. 2012;687-98.
4. Rongioletti F. Mucinosis In:Bolognia J L, Sccaffer J V, Ceroni L. Text book of dermatology , 4th edition. China, Elsevir. 2018;8,46:742-52.
5. Rebellato PR, Carbonar MB, Tabuti NI, Rastelli GJ. Case for diagnosis. Lichen myxedematosus. An Bras Dermatol. 2016;91:842-3.
6. Rongioletti F. Lichen myxedematosus (papular mucinosis):New concepts and perspectives for an old disease. Semin Cutan Med Surg. 2006;25:100-4.
7. Rongioletti F, Merlo G, Cinotti E, Fausti V, Cozzani E, Cribier B, et al. Scleromyxedema:A multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
8. Pomann JJ, Rudner EJ. Scleromyxedema revisited. Int J Dermatol. 2003;42:31-5.
9. Rongioletti F, Rebora A. Cutaneous mucinoses:Microscopic criteria for diagnosis. Am J Dermatopathol. 2001;23:257-67.
10. Rongioletti F, Rebora A. Updated classification of papular mucinosis, lichen myxedematosus, and scleromyxedema. J Am Acad Dermatol. 2001;44:273-81.
11. Rongioletti F, Zaccaria E, Cozzani E, Parodi A. Treatment of localized lichen myxedematosus of discrete type with tacrolimus ointment. J Am Acad Dermatol. 2008;58:530-2.
12. Hadj I, Gallouj S, Meziane M, Mernissi FZ. Discrete papular lichen myxedematosus:A rare entity or an under-diagnosed disease?Pan Afr Med J. 2014;19:180.
13. Concheiro J, Pérez-Pérez L, Peteiro C, Labandeira J, Toribio J. Discrete papular lichen myxoedematosus:A rare subtype of cutaneous mucinosis. Clin Exp Dermatol. 2009;34:e608-10.
14. Harris JE, Purcell SM, Griffin TD. Acral persistent papular mucinosis. J Am Acad Dermatol. 2004;51:982-8.
15. Podda M, Rongioletti F, Greiner D, Milbradt R, Rebora A, Kaufmann R, et al. Cutaneous mucinosis of infancy:is it a real entity or the paediatric form of lichen myxoedematosus (papular mucinosis)?Br J Dermatol. 2001;144:590-3.
16. Lopez-Lerma I, Fernandez-Codina A, Hilari H, Ferrer B, Selva-O’Callaghan A, Garcia-Patos V. Atypical scleromyxedema with prominent nodular lesions associated with immune thrombocytopenia:an unusual presentation. J Am Acad Dermatol. 2014;71:e158-9.
17. Zeng R, Li M, Jiang Y, Liu W. Nodular lichen myxedematosus during childhood:A case report. Pediatr Dermatol. 2014;31:e160-3.
18. Ogita A, Higashi N, Hosone M, Kawana S. Nodular-type lichen myxedematosus:A case report. Case Rep Dermatol. 2010;2:195-200.
19. Kwon OS, Moon SE, Kim JA, Cho KH. Lichen myxoedematosus with rapid spontaneous regression. Br J Dermatol. 1997;136:295-6.
20. Rongioletti F, Ghigliotti G, De Marchi R, Rebora A. Cutaneous mucinoses and HIV infection. Br J Dermatol. 1998;139:1077-80.
21. Graves MS, Lloyd AA, Ross EV. Treatment of acral persistent papular mucinosis using an Erbium-YAG laser. Lasers Surg Med. 2015;47:467-8.
22. Sperber BR, Allee J, James WD. Self-healing papular mucinosis in an adult. J Am Acad Dermatol. 2004;50:121-3.
23. Kofler H, Lipsker D, Maurer H, Burgdorf W, Requena L, Torrelo A, Zelger B, Kutzner H, Müller H. Self-healing juvenile cutaneous mucinosis:Challenging diagnosis and management. J Dtsch Dermatol Ges. 2014;12:815-7.
24. Luchsinger I, Coulombe J, Rongioletti F, Haspeslagh M, Dompmartin A, Melki I, et al. Self-healing juvenile cutaneous mucinosis:Clinical and histopathologic findings of 9 patients:The relevance of long-term follow-up. J Am Acad Dermatol. 2018;78:1164-70.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-0265-2040 http://orcid.org/0000-0002-0265-2040 |

Comments are closed.