Angiokeratoma corporis diffusum universale: A rare case report from Nepal

1Department of Dermatology and Venereology, Tribhuvan University, National Medical College, Birgunj, Nepal, 2Nepal Police Hospital, Kathmandu, Nepal
Corresponding author: Dr. Vikash Paudel
Submission: 26.06.2020; Acceptance: 01.10.2020
DOI: 10.7241/ourd.20212.18
Cite this article: Paudel V, Chudal D. Angiokeratoma corporis diff usum universale: A rare case report from Nepal. Our Dermatol Online. 2021;12(2):174-176.
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Angiokeratoma corporis diffusum universale, a clinical variant of angiokeratoma, is a generalized vascular ectasia that is associated with overlying epidermal changes rather than a true vascular neoplasm. It is the cutaneous hallmark of several rare inherited lysosomal diseases associated with specific enzyme deficiencies in the metabolism of glycoproteins. Herein, we present possibly the first case report from Nepal of angiokeratoma corporis diffusum universale in a 14-year-old female from consanguineous parents who presented herself with characteristic clinical and laboratory features of angiokeratoma corporis diffusum with progressive mental and motor developmental delay along with seizures, dystonia, and recurrent chest infections. Resource constraints limited confirmation with an enzyme assay and electron microscopy. We report this unusual case with the intention of re-emphasizing the importance of clinical evaluation in reaching a diagnosis in a resource-deficient setting.
Key words: Angiokeratoma corporis diffusum; Fabry’s disease; Fucosidosis; Nepal; Vascular ectasia
INTRODUCTION
Angiokeratomas are rare vascular ectasias with epidermal changes rather than true vascular neoplasms. These malformations clinically present themselves as single or multiple dark red to black papules, nodules, or plaques and histologically by superficial ectatic vessels with epidermal proliferation. Angiokeratoma corporis diffusum (ACD) is its diffuse involvement, typically on the lower trunk, buttocks, and thighs, usually associated with an enzyme deficiency in the metabolism of glycoproteins. For years, the term has been used interchangeably with Fabry’s disease, a systemic X-linked disease caused by a deficiency in the enzyme a-galactosidase A [1]. Angiokeratoma corporis diffusum, known to occur in several enzyme deficiency diseases involved in the metabolism of glycoproteins, such as fucosidosis, sialidosis, mannosidosis, gangliosidosis, and Kanzaki disease, is a multisystem disorder [2].
CASE REPORT
We report the case of a 14-year-old female from consanguineous parents who presented herself with recurrent episodes of fever and chest infections along with severe neurodevelopmental delay. According to the parents, the patient had had development regression since two years of age and delayed milestones in all domains. Besides, no language skills were present and vocal sounds lacked meaning. She was also showing features of seizures and myoclonic jerks. She was bedbound for six years with plasticity and generalized dystonia. A cutaneous examination revealed diffuse multiple erythematous papules and plaques of angiokeratoma over the lower back, abdomen, genitalia, and extremities (Figs. 1a and 1b). She also had coarse facies, a pigeon chest, a widened wrist elbow, and knee and ankle contractures. There was no similar family history. The clinical pictures of angiokeratoma corporis diffusum, neuroregression, spasticity, dystonia, coarse facies, dysostosis multiplex, and hepatomegaly, were suggestive of fucosidosis. Lab parameters revealed severe anemia with hemoglobin at 4.8 g/dL and a high erythrocyte sedimentation rate at 45 mm/h with normal liver, kidney, and thyroid function. Enzyme levels could not be tested because of economic constraints. Histology showed vascular ectasia with marked dilatation of papillary dermal vessels, which formed large cavernous channels, and a superficial hyperkeratotic epidermis enclosing vascular channels (Fig. 2).
 |
Figure 1: (a) The posterior trunk showing diffuse involvement of pink papules and plaques. (b) The anterior trunk showing multiple but spare dark dome-shaped papules. |
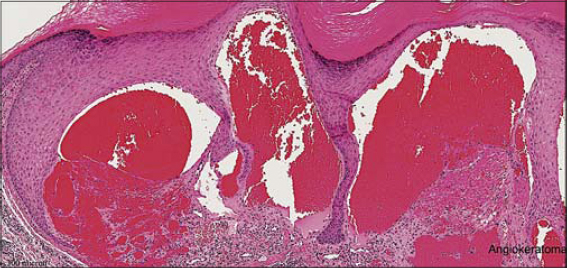 |
Figure 2: Histology of angiokeratoma showing ectasia of the upper dermal vessels with overlying epidermal hyperkeratosis. |
DISCUSSION
The term angiokeratoma describes variably-sized hyperkeratotic papules that range in color from deep red to bluish-black. Histopathologic alterations include ectasia of the upper dermal vessels with overlying epidermal hyperkeratosis [3]. Angiokeratoma corporis diffusum universale is a generalized variant of angiokeratoma, usually associated with an enzyme deficiency in the metabolism of glycoproteins [1]. Since the original independent descriptions by Anderson and Fabry in 1898, it has been recognized as a systemic disease rather than a dermatological curiosity. It is commonly seen in Fabry’s disease, fucosidosis, sialidosis, mannosidosis, gangliosidosis, or without an enzyme deficiency. Fabry’s disease is X-linked recessive, whereas the others are autosomal recessive disorders [4]. In fucosidosis, defects lead to intracellular accumulation of fucose-containing glycolipids and glycoproteins in various organs, leading to progressive mental and motor deterioration, coarse facies, recurrent infections, dysostosis multiplex, angiokeratoma corporis diffusum, visceromegaly, and seizures [5]. In our case, the typical clinical features of angiokeratoma corporis diffusum with consanguinity and neuroregression, spasticity, dystonia, coarse facies, dysostosis multiplex, and hepatomegaly were suggestive of fucosidosis. The complexity of Fabry’s disease demands a multidisciplinary therapeutic approach. There have been case reports unaided by electromicroscopic or biochemical analysis intended to re-emphasize the importance of clinical evaluation in reaching a diagnosis in a resource-deficient setting [6,7]. The role of dermoscopy has also been recently regarded as a noninvasive technique of diagnosis of angiokeratoma [8]. Ours could be the first reported case of angiokeratoma corporis diffusum from Nepal accompanied by various morbidities and systemic manifestations.
CONCLUSION
Angiokeratomas are hyperkeratotic vascular ectatic lesions that may either be localized or diffuse. There has been only a handful of reports on angiokeratoma corporis diffusum in the dermatologic literature. Thus, if a diffuse form of angiokeratoma is encountered, efforts should be made to search for systemic involvement together with a multidisciplinary approach employed.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Ivy H, Julian CA. Angiokeratoma circumscriptum. In:Stat Pearls. Treasure Island (FL):StatPearls Publishing;October 20, 2019.
2. Vadher P, Agarwal P, Mistry A, Gajjar K, Bansal N, Neazee S. Angiokeratoma corporis diffusum:an uncommon case with suspected Anderson Fabry disease. Indian Dermatol Online J. 2020;11:21-25.
3. Mittal R, Aggarwal A, Srivastava G. Angiokeratoma circumscriptum:a case report and review of the literature. Int J Dermatol. 2005;44:1031-4.
4. Bokhari SRA, Zulfiqar H, Hariz A. Fabry Disease. In:Stat Pearls. Treasure Island (FL):StatPearls Publishing;January 25, 2020.
5. Gautschi M, Merlini L, Calza AM, Hayflick S, Nuoffer JM, Fluss J. Late diagnosis of fucosidosis in a child with progressive fixed dystonia, bilateral pallidal lesions and red spots on the skin. Eur J Paediatr Neurol. 2014;18:516-9.
6. Inakanti Y, Narsimha Rao TV. A tricky man issue:Angiokeratomas of scroti. Our Dermatol Online. 2015;6:179-82.
7. Lu YY, Lu CC, Wu CS, Wu CH. Familial angiokeratoma corporis diffusum without identified enzyme defect. Indian J Dermatol Venereol Leprol. 2015;81:46-9.
8. Kamat D, Vinay K. Dermatoscopy of nonvenereal genital dermatoses:A brief review. Indian J Sex Transm Dis AIDS. 2019;40:13-9.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0001-8729-052X http://orcid.org/0000-0003-0610-3214 http://orcid.org/0000-0001-8729-052X http://orcid.org/0000-0003-0610-3214 |

Comments are closed.