Congenital erythropoietic porphyria and its rarity in Indian siblings
Sukhmani Kaur Brar1, Anjugam Alagappan 2, Balvinder Kaur Brar2
2, Balvinder Kaur Brar2
1Consultant dermatologist, Fortis medcentre, sector 11, Chandigarh India, 2Department of Dermatology, Venereology and Leprology, Guru Gobind Singh Medical College and Hospital, Faridkot, Punjab, India
Corresponding author: Dr. Anjugam Alagappan
Submission: 13.05.2020; Acceptance: 31.07.2020
DOI: 10.7241/ourd.20204.31
Cite this article: Brar SK, Alagappan A, Brar BK. Congenital erythropoietic porphyria and its rarity in Indian siblings. Our Dermatol Online. 2020;11(4):431-433.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
Sir,
Congenital erythropoietic porphyria (CEP) is a rare autosomal recessive disorder of heme biosynthesis with an estimated prevalence of 1 in 1,000,000 or less [1]. CEP is the rarest of bullous porphyrias. Only less than 200 cases have been reported worldwide, and a clinician may not happen to diagnose such a case in all their professional career [2,3].
Two male siblings, 6 and 15 years old, native residents of Uttar Pradesh, and born to a nonconsanguineous marriage presented themselves to the Department of Dermatology with a history of fluid-filled blisters in photo-exposed areas, which had healed with scarring and had been persistent from infancy, as well as reddish-brown discoloration of the urine and the teeth. No history of photosensitivity, abdominal pain, neuropsychiatric symptoms, and acute attacks was present. There were no complaints in utero or perinatally, and both siblings were born by a normal vaginal delivery from a full-term pregnancy. Milestones were normal. An examination revealed several intact blisters in the acral areas, milia, atrophic and hyperpigmented scars in photo-exposed areas, erythrodontia (Fig. 1a) in both, hypertrichosis and sclerodermatous hands in the older sibling (Figs. 1b – 1e), and less severe presentations in the younger sibling than the older (Figs. 2a – 2c). The nails showed onycholysis and dystrophy. Both showed pink fluorescence of the teeth (Figs. 3a and 3b) and the urine (Figs. 3c and 3d) in a Wood’s lamp examination and raised porphyrin levels in the plasma, urine, and feces. Histopathology of the vesicles showed a subepidermal split (Fig. 3e). Both parents and two other siblings—one male and one female—were normal, born chronologically in between the two affected siblings.
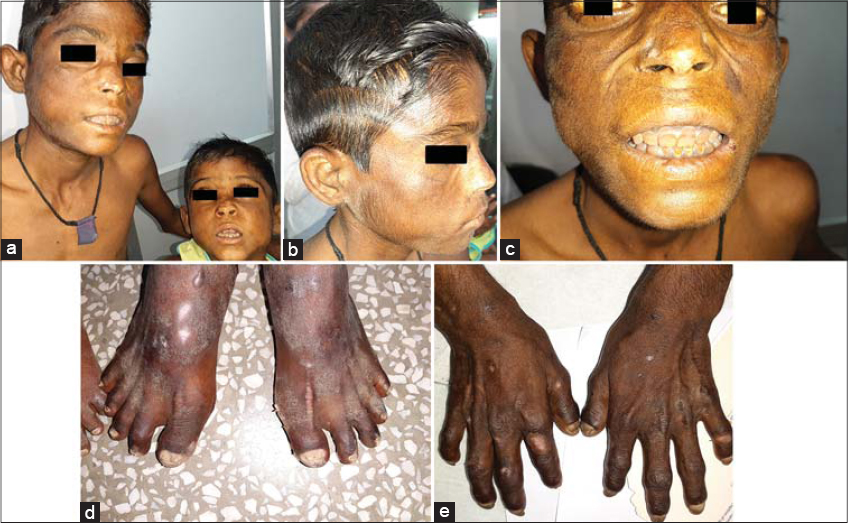 |
Figure 1: (a) Atrophic and hyperpigmented scars on the face with erythrodontia in both siblings. (b-c) Hypertrichosis on the face in the older sibling. (d-e) Sclerodermatous hands and feet with resorption of terminal phalanges in the older sibling. |
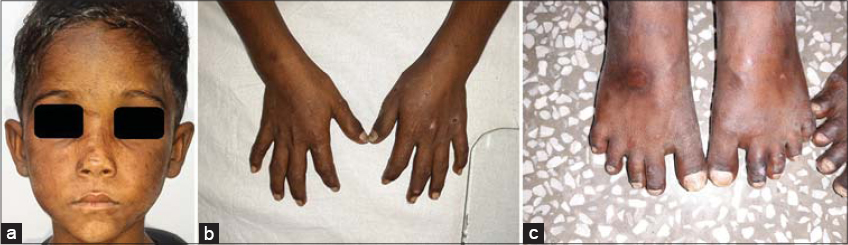 |
Figure 2: Milder involvement in the younger sibling with atrophic scars (a) on the face and (b-c) on the hands and feet. |
 |
Figure 3: (a-b) Pink fluorescence of the teeth. A brownish discoloration of the urine (c) visible to the naked eye and (d) showing pink fluorescence in a Wood’s lamp examination. (e) Histopathology of the vesicles with a subepidermal split (H&E, 100×). |
Congenital erythropoietic porphyria (CEP), also known as Gunther’s disease, is due to deficiency of uroporphyrinogen III synthase, leading to overproduction of uroporphyrinogen I and coproporphyrinogen I, which accumulate in the bone marrow, in erythrocytes, the plasma, the bones, and the teeth [3]. Its clinical spectrum is highly variable, from nonimmune hydrops fetalis to mild late-onset cases. Its onset falls typically around infancy and early childhood. One of its first clues can be a burgundy-red discoloration of the urine and diapers stained red. Cutaneous lesions begin with itching and erythema, followed by painful vesiculobullous eruptions that leave pigmented scars. In most of the other photodermatoses, inflammation is usually not severe enough to produce blisters. The presence of large amounts of porphyrins very early in life—at the time of tooth and bone development—results in erythrodontia that fluoresces under Wood’s lamp, which is almost pathognomonic of CEP [4]. Hypertrichosis can produce a werewolf appearance. Complications include hemolytic anemia, hypersplenism, hepatomegaly, bone marrow hypertrophy, pathological fractures, acral osteolysis, and ocular damage. Histopathology shows subepidermal blisters and thickened collagen bundles. Strict photoprotection remains the most important preventive measure and the first line of treatment. Treatment includes broad-spectrum sunscreen application, antioxidants, avoidance of other triggers, blood transfusions, and splenectomy in severe cases. Bone marrow and stem cell transplantation has been reported to be successful in some cases [3]. Gene therapy may provide a cure in the future. Although CEP tends to be exceptionally severe in early childhood, most patients survive into adulthood with a life expectancy of around 40 to 60 years [5].
There have been several reports of CEP from India, but almost all of them are isolated cases, and a few reports in the world literature, as in Sudanese and Pakistan siblings, but, to the best of our knowledge, none in Indian siblings. The paucity of reports of CEP in siblings in the world and Indian literature has prompted us to report these two Indian siblings.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki. The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Das S, Gunduz O, Modi Z, Modi D. A newly diagnosed South African case of congenital erythropoietic porphyria. Our Dermatol Online. 2020;11:38-40.
2. Koley S, Saoji V. Congenital erythropoietic porphyria:two case reports. Indian J Dermatol. 2011;56:94-7.
3. Raj RRM, Anuradha G, Abdul M, Riyaz MAC, Vijayavel T. Congenital erythropoietic porphyria:a rare case report. J. Indian Aca Oral Med Radiol. 2013;25:157-60.
4. Bickers DR, Frank J. The Porphyrias. In:Wolff K, Goldsmith LA, Katz SI, Gilchrest BA, Paller AS, Leffell DJ (eds). Fitzpatrick’s Dermatology in General Medicine.7th ed. New York:McGraw Hill, 2008:pp 1228-56.
5. Lefebvre T, Millot S, Richard E, Blouin JM, Lalanne M, Lamrissi-Garcia I, et al. Genetic background influences hepcidin response to iron imbalance in a mouse model of hemolytic anemia (congenital erythropoietic porphyria). Biochem Biophys Res Commun. 2019;520:297-303.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0003-0869-6906 http://orcid.org/0000-0003-0869-6906 |

Comments are closed.