A case of epithelioid angiosarcoma in a young man with chronic lymphedema
Soraya Aouali 1, Imane Alouani1, Hanane Ragragui1, Nada Zizi1,2, Siham Dikhaye1,2
1, Imane Alouani1, Hanane Ragragui1, Nada Zizi1,2, Siham Dikhaye1,2
1Department of dermatology, Mohamed 6 University Hospital of Oujda, Morocco. 2Laboratory of epidemiology, clinical research and public health, Faculty of Medicine and Pharmacy, Mohamed the first university of Oujda, Oujda, Morocco
Corresponding author: Dr. Soraya Aouali, E-mail: soraya.aouali@gmail.com
Submission: 07.09.2019; Acceptance: 07.01.2020
DOI: 10.7241/ourd.20202.13
Cite this article: Aouali S, Alouani I, Ragragui H, Zizi N, Dikhaye S. A case of epithelioid angiosarcoma in a young man with chronic lymphedema. Our Dermatol Online. 2020;11(2):165-167.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Epithelioid angiosarcoma is a rare aggressive vascular malignancy with a high recurrence and metastasis rate. Most commonly arising in the deep soft tissues while cutaneous localizations are extremely rare. We report here a case of epithelioid angiosarcoma in a young man with a specific context of lower limbs chronic lymphedema, showing clinical dermoscopic and pathology findings. Epithelioid angiosarcome is a highly malignant tumor. An early clinical and dermoscopic examination can improve the prognosis and avoid mutilating complications.
Key words: Epithelioid angiosarcoma; Lymphedema; Cutaneous localization; Dermoscopy
INTRODUCTION
Epithelioid angiosarcoma is a highly aggressive endothelial cell malignancy, most commonly arising in the deep soft tissues, with a minority falling into the visceral and cutaneous categories. It’s a rare variety that represents less than 1% of all sarcomas [1].
Angiosarcoma can arise de novo (primary angiosarcoma) or secondary to either local irradiation or in patients with long-standing lymphedema (secondary angiosarcoma).
This report provides clinical, dermoscopic and pathology findings in an epithelioid cutaneous angiosarcoma occurring in a specific context of lower limbs chronic lymphedema.
CASE REPORT
A 30 years-old man with lower limbs chronic lymphedema, progressively increasing, was referred to our department for a right lower ulcerative tumor, evolving for 3 months prior to his referral. The physical examination revealed stage 2 lymphedema of both lower limbs and a rounded ulcero-budding tumor with raised edges, measuring 13×10 cm in the pretibial region of his right leg (Fig. 1). Dermoscpic examination showed structureless patchy purpule and orange-red areas, with white-pearly stripes (Fig. 2). Lymph nodes examination reveals 2 inguinal nodes. Diagnosis of verrucous squamous cell carcinoma, sarcoma or atypical mycobacterial infection were suspected. A skin biopsy was performed and histopathology showed an extensive infiltrate of the dermis and hypodermis with a malignant proliferation of rounded cells with an atypical vesicular nuclei, a high mitotic index and foci of tumor necrosis. These atypical cells strongly expressed CD31 and EGR, typical of epithelioid sarcoma. While immunostaining for CD34, cytokeratin and EMA were negative. These morphological features and immunochemistry study confirmed the diagnosis of a high grade epithelioid angiosarcoma (grade 3 of the FNCLCC classification). No metastatic localizations were found after performing a computed tomography scan. Due to its aggressive nature, high metastasis and recurrence rate, leg amputation with lymph node dissection associated to adjuvant radiation therapy was decided.
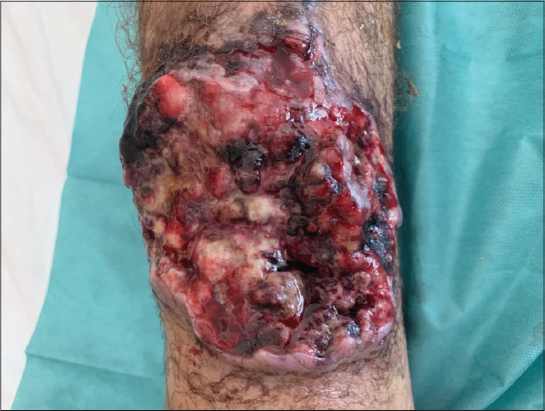 |
Figure 1: Rounded ulcero-budding tumor with raised edges in the pretibial region. |
 |
Figure 2: Dermoscopic findings: structureless patchy purpule and orange-red areas, with white-pearly stripes. |
DISCUSSION
We report a case of epithelioid angiosarcoma in the setting of chronic lymphedema.
Why chronic lymphedema can lead to angiosarcoma is unclear and controversial, it can be explained by the fact that chronic lymphedema causes local immunodeficiency, therefore indirectly promoting oncogenesis. According to Ruocco et al. when the local mechanisms of immune surveillance begin to fail, the lymphedematous region becomes an immunologically vulnerable area, predisposed to malignancy including vascular tumors such as angiosarcoma [2]. Secondary angiosarcoma may involve the skin, deep soft tissues, or deep organs.
Cutaneous secondary angiosarcoma can develop in a context of chronic lymphedema (Stewart- Treves syndrome). More than 200 cases of cutaneous secondary angiosarcoma have been reported in the context of lymphedema of the upper extremity, occurring 10 to 20 years following radical mastectomy [3]. In contrast, secondary angiosarcoma occurring with congenital lymphedema is even more rare. The first case was reported by Kettle in 1918 [4] since then, less than 30 case were reported in the literature, each occurring in congenital lymphedema.
The clinical presentation of angiosarcoma described usually relates to its common presentation in the upper extremity. This includes initially either a palpable subcutaneous mass or a poorly healing eschar with recurrent bleeding. In later advanced stages, multiple red-blue macules or nodules develop and may become polypoid, with ulceration and necrosis complicating the late stage of some lesions.
Dermoscopic examination can help the clinician by revealing the classic colors of vascular lesions, graduation of red, purple, and blue. Various color gradiation may be an important dermoscopic feature of cutaneous angiosarcoma, In addition, cutaneous angiosarcoma is characterized by the absence of well-defined vascular structures with patchy whitish and purpule veil [5], similar to the features found in our case.
Histological examination shows predominant poorly differentiated epithelioid clear cell morphology. Epithelioid appearance is more common in angiosarcomas arising in deep soft tissues than those developed in skin. The positivity of a panel of vascular immunohistochemical markers, including CD31, CD34, and ERG, usually confirms the diagnosis [6].
The molecular pathogenesis and cytogenetics of cutaneous secondary angiosarcoma in the context of chronic lymphedema are poorly characterized. The most frequent changes were high-level amplifications on chromosome 8q24.21 [7]. In a series of 25 cases, Mentzel et al. confirmed the presence of an amplification of c-MYC in all cases of secondary angiosarcoma studied by FISH. Immunostaining for c-MYC has been developed and might help to distinguish angiosarcoma from atypical vascular proliferation in difficult cases [8].
Therapeutic options for epithelioid angiosarcoma include surgery, radiotherapy, and chemotherapy, singly or in combination. The small number of reported cases to date precludes determination of the optimum treatment regime at this stage, although where possible, wide excision is recommended. The need for adjuvant therapy is determined on an individual basis.
The overall prognosis of epithelioid angiosarcoma is poor, with a high rate of local recurrence and distant metastasis, regardless of the chosen treatment.
CONCLUSION
In summary, epithelioid angiosarcoma is a rare, aggressive vascular malignancy with a high recurrence and metastasis rate. An early dermatological evaluation and appropriate dermoscopic examination may help avoid a delayed diagnosis, to get a better prognosis with less mutilating complications.
REFERENCES
1. Rouhani P, Fletcher CDM, Devesa SS, Toro JR. Cutaneous soft tissue sarcoma incidence patterns in the U.S.:an analysis of 12,114 cases. Cancer. 2008;113:616–27.
2. Ruocco V, Schwartz RA, Ruocco E. Lymphedema:an immunologically vulnerable site for development of neoplasms. J Am Acad Dermatol. 2002;l47:124–7.
3. Fayette J, Martin E, Piperno-Neumann S, Le Cesne A, Robert C, Bonvalot S, et al. Angiosarcomas, a heterogeneous group of sarcomas with specific behavior depending on primary site:a retrospective study of 161 cases. Ann Oncol. 2007;18:2030–6.
4. Offori TW, Platt CC, Stephens M, Hopkinson GB. Angiosarcoma in congenital hereditary lymphoedema (Milroy’s disease)—diagnostic beacons and a review of the literature. Clin Exp Dermatol. 1993;18:174–77.
5. Cozzani E, Chinazzo C, Ghigliotti G, Pastorino C, Gasparini G, Parodi A. Cutaneous angiosarcoma:the role of dermoscopy to reduce the risk of a delayed diagnosis. Int J Dermatol. 2018;57:996-7.
6. Shavit E, Alav A, Limacher JJ, Sibbald RG Angiosarcoma complicating lower leg elephantiasis in a male patient:An unusual clinical complication, case report and literature review. SAGE Open Med Case Rep. 2018;6:2050313X18796343.
7. Itakura E, Yamamoto H, Oda Y, Tsuneyoshi M, Detec- tion and characterization of vascular endothelial growth factors and their receptors in a series of angiosarcomas. J Surg Oncol. 2008;9774–81.
8. Fernandez AP, Sun Y, Tubbs RR, Goldblum JR, Billings SD. FISH for MYC amplification and anti-MYC immuno- histochemistry:useful diagnostic tools in the assessment of secondary angiosarcoma and atypical vascular proliferations. J Cut Pathol. 2012;39234–42.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0001-7687-0158 http://orcid.org/0000-0001-7687-0158 ResearcherID: K-2386-2016 ResearcherID: K-2386-2016 |

Comments are closed.