|
Get Citation
|
|
|
Lubkov A, Uraga V, Briones M-C, Uraga E. Cornelia de Lange syndrome and psoriasis: Report of a case. Our Dermatol Online. 2018;9(1):38-40. |
|
|
Download citation file:
|
Cornelia de Lange syndrome and psoriasis: Report of a case
Andrea Lubkov1, Veronica Uraga1, Maria-Cecilia Briones2, Enrique Uraga1,2
1Deparment of Dermatology, Hospital “Luis Vernaza”, Guayaquil, Ecuador; 2Deparment of Dermatology, Centro Privado de Piel “Dr. Enrique Uraga Peña”, Guayaquil, Ecuador
Corresponding author: Dr. Andrea Lubkov, E-mail: andrealubkov@gmail.com
Submission: 08.08.2017; Acceptance: 13.10.2017
DOI: 10.7241/ourd.20181.11
ABSTRACT
Cornelia de Lange syndrome was first reported in 1916 and it is characterized by growth deficiency, psychomotor delay and unique facial expression. Its incidence is 1:45.000 live births, being an infrequently reported entity. We present the case of a 15 years old girl with Cornelia de Lange syndrome associated with Psoriasis.
Key words: Cornelia de Lange; Psoriasis; Syndrome
INTRODUCTION
Cornelia de Lange syndrome is a multi systemic disease, first reported in 1933 by Cornelia Catharina de Lange, who described two cases and named it “typus degenerativus amstelodamensis”. A similar case was described in 1916 by Winfred Brachmann, and then renamed it as “Brachmann-deLange syndrome”. It is characterized by growth deficiency and psychomotor delay, feeding and behavioral difficulties [1,2].
The incidence is variable, ranging from 1:10.000 to 1:30.000 live births. Some cases are sporadic, related to genes SMC1A, SMC3, RAD21 and HDAC8 on chromosome X, but also some of them are autosomic dominant, related to gene NIPBL on chromosome 5, responsible for 50% of the cases. This syndrome has a wide spectrum of manifestations that includes neurological, endocrinological, muscle-skeletal and cutaneous abnormalities [2].
CASE REPORT
A 15 years old female patient, with Cornelia de Lange syndrome, presents since 9 years old relapsing erythematous descamative plaques, some of them with a circinate or nummular pattern, disseminated but more intense in trunk and extremities, with severe pruritus (Figs. 1 and 2). The girl’s mother referred that she presented at birth low weight, congenital heart disease (paten foramen oval presumed due to its spontaneous closure), seizures and hip dislocation, that led to the diagnosis of Cornelia de Lange syndrome.
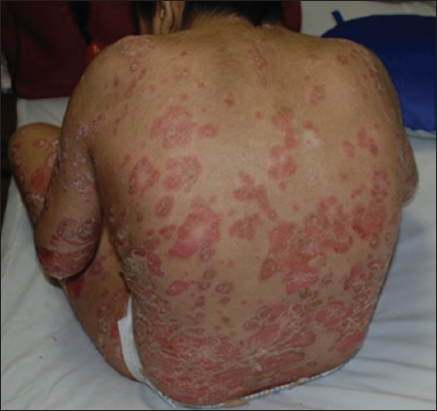
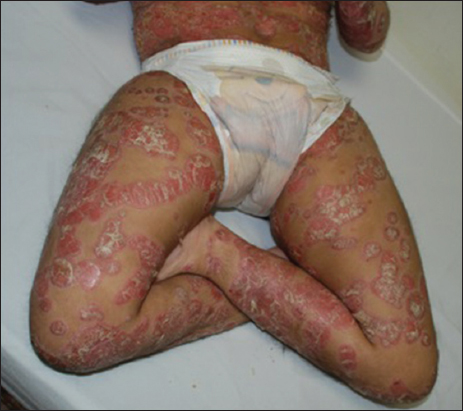
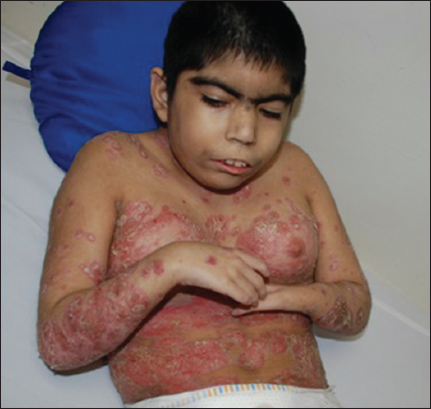
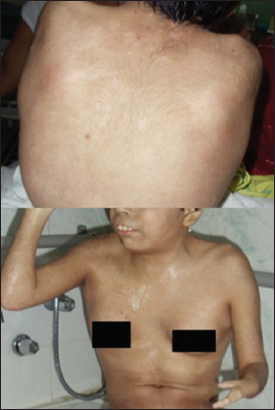
In the physical exam, we noticed weak cry, psychomotor delay, short stature, synophrys, low hairline, hirsutism, down-turned angles of the mouth, low-set ears, and muscle-skeletal abnormalities (Fig. 3). A histopathological diagnosis of psoriasis was made, as we examined our therapeutic options. Treatment with topical clobetazol and coal tar was established showing complete resolution of the lesions after one month of treatment (Fig. 4).
DISCUSSION
Cornelia de Lange syndrome (CdLS) is also known as Brachmann de Lange syndrome, it’s characterized by a wide phenotypical spectrum, which makes it easy to diagnosis, although some cases remained undiagnosed years after birth [2]. The genes implicated in its pathophysiology are SMC1A, SMC3, RAD21 and HDAC8 that codify proteins of the cohesine complex [3].
These patients are characterized by short stature, and psychomotor delay. Mean stature for men is 156 cm and 131 cm for women. Some cases present central obesity. Multiple associations have been described, including gastrointestinal abnormalities, as gastroesophageal reflux between the most common [4]. Other alterations may be present including ophthalmologic, cardiovascular, and urinary [5,6].
Facial features include hirsutism, synophrys, long and thick eyelashes, low-set ears, broad nasal bridge, thin superior lip, down-turned angles of the mouth, and micrognatia [7,8]. Hirsutism is a common feature, although some patients present alopecia. Cutis marmorata and cutis verticis gyrata have been described [5]. Xerosis and hypoplasic borders of the fingers are observed, the latter often leading to erasure of finger prints [8–10]. According to its phenotypic expression, CdLS can be classified in three groups (Table 1); mild, moderate and severe disease, due to the extremities, growth and psychomotor affection [11–13].

Another classification divides it in [1]:
- Classical phenotype: Characteristic facial and skeletal changes.
- Mild phenotype: Mild characteristic facial and skeletal changes.
- CdLS-like: Similar phenotypic expression but related to chromosomic or teratogenic affections.
Diagnosis is made when [1]:
- Positive affected gene
- Facial features and two growth, developmental or behavioral criteria
- Facial features and, one growth, developmental or behavioral criteria, and two additional criteria.
A severity score system has been established, that includes physical and psychomotor development, which divides them according to its classification in severe involvement (>22 points), moderate involvement (15-22 points), mild involvement (<15 points) (Table 2) [14]. Our patient score was 25 points.

Several case reports have associated CdLS with other pathologies, although dermatologic associations are infrequently reported. In the literature at our disposal we found an association with rosacea in a 16 years old female [15], and Uleritema Ofriogenes in a 17 years old female [16]. Our patient represents the second case of CdLS associated with psoriasis [17].
CONCLUSION
Although psoriasis is a well known and commonly reported disease, its incidence in CdLS is extremely low. This association is very rare, to the best of our knowledge; there is only one previous report in a Pakistani girl. Our case represents a therapeutic challenge due to the limited options available for this specific case. We had a rapid response with only topic corticoids and coal tar.
REFERENCES
1. Mikołajewska E. Interdisciplinary therapy in Cornelia de Lange syndrome – review of the literature. Adv Clin Exp. 2013;22:571–7.
2. Vall J. Cornelia de Lange syndrome. Med J Aust. Am Soc Surg Hand. 1978;1:335.
3. Deardorff MA, Kaur M, Yaeger D, Rampuria A, Korolev S, Pie J, et al. Mutations in Cohesin Complex Members SMC3 and SMC1A Cause a Mild Variant of Cornelia de Lange Syndrome with Predominant Mental Retardation. Am J Hum Genet. 2007;80:485–94.
4. Castronovo P, Delahaye-Duriez A, Gervasini C, Azzollini J, Minier F, Russo S, et al. Somatic mosaicism in Cornelia de Lange syndrome:A further contributor to the wide clinical expressivity? Clin Genet. 2010;78:560–4.
5. Collis L, Moss J, Jutley J, Cornish K, Oliver C. Facial expression of affect in children with Cornelia de Lange syndrome. J Intellect Disabil Res. 2008;52:207–15.
6. Koch A, Eisig S. Syndromes with unusual facies. Atlas Oral MaxillofacSurgClin North Am [Internet]. Elsevier Inc. 2014;22:205–10.
7. He Q, Johnston J, Zeitlinger J, City K, City K. HHS Public Access. 2015;33:395–401.
8. Macchini F, Fava G, Selicorni A, Torricelli M, Leva E, ValadèA. Barrett’s esophagus and Cornelia de Lange Syndrome. Acta Paediatr. 2010;99:1407–10.
9. Humberto D, Fernando J, Daniel M. Sindrome Cornelia de Lange. Rev Chil Pediatr.1980;51:55-8.
10. Salazar FN, De Lange C, Opitz JM, Laurence KM, Ptacek LJ, Jervis GA, et al. Dermatological Manifestations of the Cornelia de Lange Syndrome. Arch Dermatol Am Med Assoc. 1966;94:38.
11. Gillis LA, McCallum J, Kaur M, DeScipio C, Yaeger D, Mariani A, et al. NIPBL mutational analysis in 120 individuals with Cornelia de Lange syndrome and evaluation of genotype-phenotype correlations. Am J Hum Genet Elsevier. 2004;75:610–23.
12. Recavarren AM, Araníbar DL, Ureta LP, Castillo TS, Tellerías CL. Síndrome de Cornelia de Lange. Rev Chil Dermatol. 2008;3:233–6.
13. Gil MC, Ribate MP, Ramos FJ. Síndrome de Cornelia de Lange. Protoc Diagn Ter Pediatr. 2010;1:1-12.
14. Malik LM, Khan GA, Azfar NA, Jahangir M. Case Report Cornelia de Lange Syndrome – a cause of hypertrichosis in children:Case report and review of literature. J Pak Assoc Dermatol. 2011;21:211–4.
15. Eghlileb AM, Finlay AY. Granulomatous rosacea in Cornelia de Lange syndrome. Indian J Dermatol Venereol Leprol. 2009;75:74–5.
16. Flórez A, Fernández-Redondo V, Toribio J. Ulerythemaophryogenes in Cornelia de Lange syndrome. Pediatr Dermatol.2016;19:42–5.
17. Shahzad A, Suhail Pal S, Khurshid K. Cornelia De Lange Syndrome with generalized pustular psoriasis:A rare coexistence. J Pakistan Assoc Dermatologists. 2005;15:281–4.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.