|
Get Citation
|
|
|
Bains P. Classification criteria of systemic sclerosis: Journey so far. Our Dermatol Online. 2017;8(2):220-223. |
|
|
Download citation file:
|
Classification criteria of systemic sclerosis: Journey so far
Pooja Bains
Department of Skin and Venereology, SGRDIMSR, Amritsar, Punjab, India
Corresponding author: Dr. Pooja Bains, E-mail: pjdhawan76@gmail.com
Submission: 20.06.2016; Acceptance: 16.08.2016
DOI: 10.7241/ourd.20172.61
ABSTRACT
Systemic sclerosis (SSc) is a rare connective tissue disease of obscure etiology with heterogenous manifestations. In order to understand this disease and improve patient prognosis, various classification criteria have been proposed over time but as these criteria lack sensitivity and specificity, early identification of patients is still a difficult task. The improved understanding of disease pathogenesis has led to reanalysis and updating of existing classification criteria for SSc. Researchers are emphasising on the importance of nail fold capillaroscopy and SSc related auto antibodies as significant criteria for early diagnosis of SSc. These newer classification criteria need to be extensively used and validated before they gain universal acceptance.
Key words: Heterogenous; Nail fold capillaroscopy; Autoantibodies
INTRODUCTION
Systemic Sclerosis is a multi system, multistage connective tissue disease characterized by vasculopathy, fibrosis and degenerative changes in the skin and internal organs and production of autoantibodies [1]. This disease may involve one or many internal organs including heart, lungs, gastrointestinal tract. The spectrum of manifestations and prognosis of Systemic Sclerosis (SSc) is variable. Early identification of SSc patients is of great importance to delay the development of complications and to screen patients for severe organ involvement. In the last two decades, there has been a better understanding of the natural course of the disease and a remarkable progress in the diagnostic tests for Systemic sclerosis [2].
The diagnostic criteria for SSc are lacking, although there are several classification and subset classification criteria proposed for SSc which aid in diagnosis [3]. The Classification criteria are standardised criteria which help in differentiating patients with the disease in question from those without the disease. The basic utilization of classification criteria is for clinical trials and research studies but since they closely mimic the diagnostic criteria, they can be used as a basic tool in identifying patients in early stages of Systemic Sclerosis. The patients classified as having SSc are a subset of patients being diagnosed as having SSc, with diagnosis being more sensitive [4]. The classification criteria serve as important guidelines for differentiating SSc from various overlapping diseases on the basis of clinical and serological parameters. In this article, an attempt has been made to review the history of classification and subsetting of SSc with special emphasis on the recently proposed EULAR classification criteria.
In 1978, the first classification of SSc was proposed by Barnett [5], according to which there were three subsets: Type I, with skin changes involving only the fingers; Type II, with sclerosis limited to the forearms; and Type III, with diffuse skin involvement. A couple of years, after this attempt by Barnett, the first standard classification criteria for Systemic Sclerosis were developed by the American College of Rheumatology (ACR) in 1980 [6]. The ACR criteria have the advantage of being well researched and validated in a large population of patients along with 92% sensitivity and 96% specificity [7]. These are often used to diagnose patients of SSc [3]. According to these criteria, patient should have either one major or two out of three minor criteria.
- Major criteria: Scleroderma proximal to the digits, affecting limbs, face, neck or trunk
- Minor criteria: At least two minor criteria out of the three:
- sclerodactyly
- digital pitted scarring
- bilateral basal pulmonary fibrosis
Drawbacks of ACR criteria:
- The ACR criteria do not deal with the heterogeneity of SSc [5]
- These criteria were developed by using patients with long standing disease so chances of missing early Systemic Sclerosis is high [8]
- These criteria are insufficient to diagnose cases with limited form of SSc [2]
- The characteristic nail fold changes of SSc, which are an important early feature in SSc, are not included in these criteria [9]
- The diagnostic tests for autoantibodies which aid early diagnosis, have improved over the years [9].
- Less sensitivity [7].
The second classification criteria were proposed by LeRoy in 1988 [10]. The main highlight of these criteria was that it differentiated the two main subsets of Systemic Sclerosis: Diffuse form of SSc (dcSSc) and Limited Cutaneous form of SSc (lcSSc.). The LeRoy classification is shown in Table 1 [6]. The main advantage of this classification is its ease of use in everyday practice and wide acceptance [7]. The drawback is that this classification is highly exclusive. There is an unsettling dilemma of whether the diffuse and limited forms are different diseases or represent different phenotypes of the same disease [5]. Another major drawback is that patients with early disease, without or with minimal skin changes and no internal organ involvement, do not fit in this classification [3].
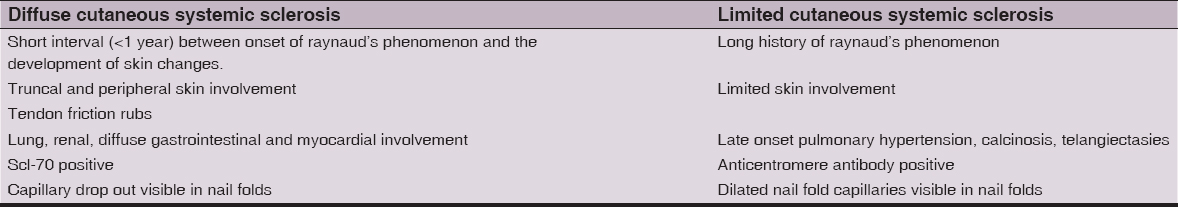
The LeRoy classification was revised and a modified classification was proposed by LeRoy & Medsger in 2001 [5]. Table 2 shows the LeRoy & Medsger classification [10]. This classification differentiated Early SSc using nail fold capillaroscopy and SSc related autoantibodies and included an additional early or limited form of scleroderma, lSSc, to supplement the previously recognized lcSSc and dcSSc forms [5]. According to LeRoy and Medsger, adding nailfold capillary findings and anticentromere serology, improved the sensitivity of ARA classification, highlighting the key role played by these two features [9]. Despite its advantages, this classification has not been validated [3]. The confusion in differentiating the two forms lSSc and lcSSc is another drawback of this classification [5].

In 2004, Maricq and Valter proposed a further set of classification criteria of SSc as shown in Table 3 [5]. This classification tries to subclassify the disease and incorporates new diagnostic technologies. The main drawback is that it has not been validated externally nor has it been tested in control population [7]. These criteria are quite complex and are not easy to apply making their wide acceptance difficult [5].

The need for revised criteria for SSc arose because of various reasons [8]:
- The mere absence of cutaneous involvement does not exclude the diagnosis of SSc as it is a multiorgan disease with variable internal organ manifestations.
- The immunological and vascular changes occur early in disease but are not given significance in the previous classifications.
- Since 1980, significant advances have occurred in the understanding of disease pathogenesis, resulting in need for newer criteria.
- No classification criteria so far has received universal acceptance.
The revised SSc classification criteria should satisfy the following requirements [7]:
- They should include the complete spectrum of SSc and should apply to patients that are early as well as late in the disease process.
- They should include vascular, immunologic, and fibrotic manifestations.
- They should be feasible in daily clinical practice and clinical studies.
- They should be as close as possible to items used for diagnosis in clinical practice.
- They should be more sensitive and specific than the previous criteria.
The newest criteria which has been proposed in 2013 is the ACR/EULAR classification. This classification includes one definitive criteria which is sufficient to make diagnosis of SSc and seven criteria with point system which are used if definitive criteria is not fulfilled. The total score is determined by adding the maximum weight (score) in each category. Patients with a total score of 9 are classified as having definite SSc. The ACR/EULAR 2013 criteria are shown in Table 4 and the definitions of items/sub-items for these criteria are given in Table 5 [4].
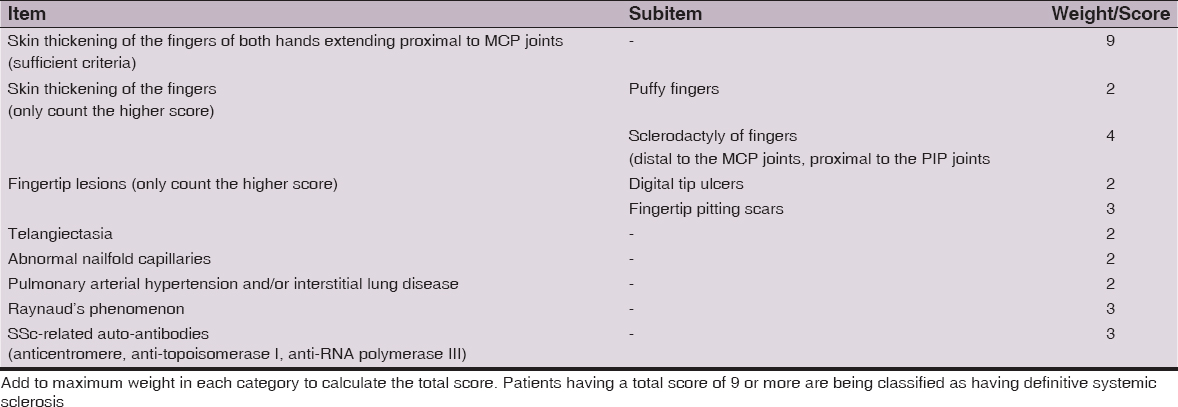

Advantages of ACR/EULAR criteria [3]:
- These have greater sensitivity and specificity than the previous criteria. Sensitivity and specificity in the validation sample for EULAR criteria is 0.91 and 0.92 in comparison to 0.75 and 0.72 for the 1980 ACR classification criteria.
- These criteria perform well even in patients with early SSc (less than 3 years).
- These criteria combine the significant points of previous all classifications along with new criteria.
- These criteria include newer advances in the diagnostic techniques like specific serum autoantibodies.
- The present classification does not require any computing device, hence can be easily used in individual subjects.
- ACR/EULAR criteria have an advantage as it uses two standards for testing and validation of the proposed system.
CONCLUSION
To facilitate early diagnosis in SSc, improving patient care and prognosis, there is need for a validated and well accepted set of classification criteria for SSc. There is still lack of an incontrovertible test or criteria for SSc. Till that time, the classification criteria for SSc remains an evolving issue, needing more scientific research.
REFERENCES
1. Goodfield MJD, Jones SK, Veale DJ. The Connective Tissue Diseases.In: Burns T, Breathnach S, Cox N,Griffiths C, editors. Rook’s Textbook of Dermatology. 8th ed.Oxford: Wiley-Blackwell;2010.p.51.87.
2. Kowal Bielecka O, Bielecka M, Kowal K. Recent advances in the diagnosis and treatment of systemic sclerosis. Pol Arch Med Wewn. 2013;123:51-8.
3. Vonk MC, van den Hoogen FH, van Riel PL, Valentini G. What does the clinician need to improve patient care in systemic sclerosis? Ann Rheum Dis. 2007;66:1129-31.
4. Van den Hoogen F, Khanna D, Fransen J, Johnson SR, Baron M, Tyndall A et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against Rheumatism collaborative initiative. Rheumatology/European League against Rheumatism collaborative initiative. Arthritis Rheum. 2013;65:2737-47.
5. Wollheim FA. Classification of systemic sclerosis. Visions and reality. Rheumatology (Oxford). 2005;44:1212-6.
6. Goodfield MJD, Jones SK, Veale DJ. The Connective Tissue Diseases.In: Burns T, Breathnach S, Cox N, Griffiths C, editors. Rook’s Textbook of Dermatology. 8th ed.Oxford: Wiley-Blackwell;2010.p.51.91.
7. Walker JG, Pope J, Baron M, Leclercq S, Hudson M, Taillefer S et al. The development of systemic sclerosis classification criteria. Clin Rheumatol. 2007;26:1401-9.
8. Fransen J, Johnson SR, van den Hoogen F, Baron M, Allanore Y, Carreira PE. Items for developing revised classification criteria in systemic sclerosis: Results of a consensus exercise. Arthritis Care Res (Hoboken). 2012;64:351-7.
9. Sierakowski S, Gińdzieńska-Sieśkiewicz E, Kowal-Bielecka O. The importance of early diagnosis of systemic sclerosis. Rocz Akad Med Bialymst. 2005;50 Suppl 1:232-3.
10. LeRoy EC, Black C, Fleischmajer R, Jablonska S, KriegT,MedsgerTAJr, et al. Scleroderma (systemic sclerosis): classification,subsets and pathogenesis. JRheumatol. 1988;15:202–5.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.