|
Get Citation
|
|
|
Diallo M, Touré M, Diatta BA, Diop A, Ndiaye M, Diop MT, Seck B, Niang S. Maladie de Rosaï-Dorfman a presentation cutanee tumorale. [Rosaï-Dorfman disease with tumoral skin lesions]. Our Dermatol Online. 2017;8(suppl. 1):40-43. |
|
|
Download citation file:
|
Maladie de Rosaï-Dorfman a presentation cutanee tumorale
[Rosaï-Dorfman disease with tumoral skin lesions]
Moussa Diallo1, Maïmouna Touré2, Boubacar Ahy Diatta1, Assane Diop1, Maodo Ndiaye1, Mame Téné Diop1, Birame Seck1, Suzanne Niang1
1Department of Dermatology, Aristide Ledantec University Hospital of Dakar, Senegal, 2Department of Internal Medecine, Aristide Ledantec University Hospital of Dakar, Senegal
Corresponding author: Prof. Moussa Diallo, E-mail: moussante@hotmail.com
Submission: 11.09.2017; Acceptance: 28.10.2017
DOI: 10.7241/ourd.2017s.10
ABSTRACT
Rosai-Dorfmann disease manifests on the skin commonly as papules, nodules or rarely as infiltrated plaques. Authors report the case of a 27 year-old-man, presenting diffuse, budding and pediculated tumoral skin lesions associated with superficial and deep lymphadenaopathies and fever. The histopathological examination showed caracteristic features of emperipolesis. The Rosaï-Dorfman disease is remarkable in our case by its tumoral and profus presentation, as well as its pseudo-xanthomatous aspect.
Key words: Rosai-Dorfman disease, Emperipolesis, Lymphadenopathies
RÉSUMÉ
Les lésions cutanées de la maladie de Rosai-Dorfman sont le plus souvent à type de papules, nodules ou plus rarement de plaques infiltrées. Les auteurs rapportent l’observation d’un homme de 27 ans, présentant des lésions tumorales cutanées bourgeonnantes, pédiculées, diffuses, associées à des polyadénopathies superficielles et profondes fébriles, avec des images caractéristiques d’empéripolèse à l’histologie. La maladie de Rosaï-Dorfman est remarquable dans notre observation par sa présentation cutanée tumorale et profuse, ainsi que l’aspect pseudo-xanthomateux.
Mots clés: Maladie de Rosai-Dorfman, Empéripolèse, Lymphadénopathies
INTRODUCTION
La maladie de Rosai-Dorfman (MRD) ou histiocytose sinusale avec lymphadénopathie est une histiocytose non langerhansienne rare, acquise, d’étiologie encore inconnue [1]. Elle est habituellement caractérisée par des polyadénopathies diffuses, surtout cervicales associées à une fièvre [2]. Les localisations extra-ganglionnaires sont possible et la peau est l’organe le plus souvent atteint. Les lésions cutanées sont le plus souvent à type de papules, nodules ou plus rarement de plaques infiltrées [3]. Nous rapportons l’observation d’une localisation cutanée d’une MRD particulière par le caractère tumoral et profus des lésions.
OBSERVATIONS
Un homme âgé de 27 ans, originaire de la Guinée Conakry, sans antécédent notable, a été admis pour des lésions papulo-nodulaires et tumorales cutanées diffuses, associées à des polyadénopathies cervicales et une fièvre.
Le début de sa maladie remonte à 4 ans, marqué d’abord par une fièvre à prédominance vespèro-nocturne associée à des frissons sans sueur, suivies 2 mois plus tard de l’apparition de lésions papulo-nodulaires, asymptomatiques, augmentant progressivement en taille et en nombre. Parallèlement, il a présenté des adénopathies cervicales, axillaires et inguinales.
Ce tableau clinique, associé à une intradermoréaction à la tuberculine (IDRT) positive, avait motivé un traitement antituberculeux à Conakry mais sans succès. L’examen histopathologique ganglionnaire effectué toujours en Guinée avait suggéré un possible lymphome, motivant une chimiothérapie à base de CHOP (cyclophosphamide, hydroxyadriamycine, oncovin, prednisone) pendant 4 mois avec une amélioration transitoire, suivie d’une augmentation de volume des lésions.
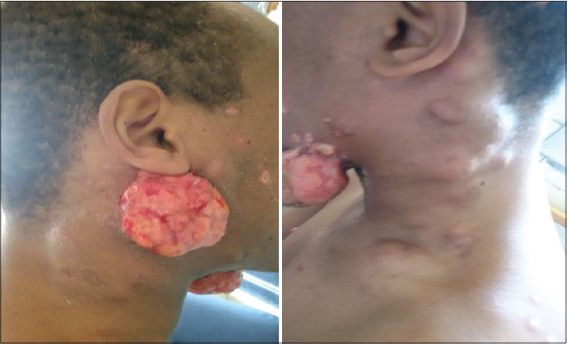
A l’admission, l’examen clinique mettait en évidence 4 masses tumorales de 4 à 6 cm de diamètre, bourgeonnantes, pédiculées au niveau des régions sous maxillaire gauche et parotidienne droite (Fig. 1a). En plus, il existait des lésions papulo-nodulaires jaunâtres, télangiectasiques, de taille variable de 0,5 à 2cm, de consistance molle, disséminées sur le visage, le tronc, le dos, les zones pilaires (cuir chevelure, narines et pubis). L’examen mettait aussi en évidence des polymacroadénopathies fixées, indolores, fermes, au niveau des aires cervicales, axillaires et inguinales (Fig. 1b). Le reste de l’examen clinique en particulier ophtalmologique, neurologique et ORL était normal.
Les examens biologiques montraient une anémie hypochrome microcytaire inflammatoire à 8,3 g/dl, une thrombocytose à 474.000/mm3 et un syndrome inflammatoire non spécifique (CRP à 48 mg/l, hypera2globulinemie à 9,77g/L) Il existait une gammapathie polyclonale à 34,97g/l à l’électrophorèse des protéines sériques.
L’échographie abdominale objectivait des polyadénopathies de siège hilaire hépatique, splénique et lombo-aortique. Une cyto-ponction ganglionnaire montrait de nombreux macrophages, des polynucléaires neutrophiles, des lymphocytes et des plasmocytes.
L’examen histopathologique d’un nodule cutané montrait un dense infiltrat dermique, composé de grands histiocytes à cytoplasme large et clair, disposés en nappe, montrant des images d’empéripolèse et associés à de rares cellules géantes de Touton, des plasmocytes, des lymphocytes et quelques neutrophiles (Fig. 2). Les colorations par le Ziehl et le PAS étaient négatives.

Le patient était mis sous Prédnisone à raison de 1,5 mg/kg avec une fonte spectaculaire des adénopathies et une régression des lésions cutanées au bout de 3 semaines. Le patient est suivi, sans récidive encore depuis 2 mois.
DISCUSSION
La maladie de Rosai-Dorfman est une histiocytose non langerhansienne, inflammatoire, rare [4–6]. Elle est observée préférentiellement chez l’adulte jeune d’âge moyen de 20,6 ans avec une légère prédominance masculine (3 hommes pour 2 femmes) [5]. La prédominance chez les noirs africains évoquée par certains auteurs reste cependant contestée [7]. Toutefois, la rareté des cas rapportés en Afrique noire pourrait aussi être en rapport avec une méconnaissance de cette affection rare par les praticiens dans cette région. A ce jour, les plus grandes séries concernent surtout des patients occidentaux ou d’origine asiatique [6]. Dans sa forme classique, la MRD se manifeste typiquement par un tableau de polyadénopathies non inflammatoires fébriles [1,2]. Dans 43% des cas, une localisation extra-ganglionnaire cutanée, ORL, osseuses, orbitaires ou neurologique peut être observée [7,8]. L’atteinte cutanée, observée dans 16% des cas, est la plus fréquente des localisations extra-ganglionnaires [7]. Elle peut parfois être révélatrice et plus rarement être isolée. La présentation clinique cutanée est polymorphe et peu spécifique, rendant souvent le diagnostic difficile. Typiquement, il s’agit de papules et de nodules de quelques millimètres ou centimètres, en nombre variable ou rarement de plaques infiltrées, siégeant préférentiellement dans la région cervico-céphalique et la partie haute du tronc [3,5–7]. Des lésions pustuleuses acnéiformes ont aussi été rarement rapportées [9]. Dans notre observation, la présentation de la MRD était remarquable par le caractère tumoral et profus des lésions cutanées, ainsi que leur couleur franchement jaunâtre, pseudo-xanthomateuse. Cette présentation particulière avait certainement aussi contribué au retard diagnostique fréquent au cours de cette affection rare et peu connue des praticiens. Des délais diagnostiques de 19 ans ont d’ailleurs été rapporté même dans les pays développés [10]. En effet, le diagnostic est généralement porté tardivement, souvent après plusieurs biopsies ou relecture des lames histologiques. L’orientation diagnostique par les cliniciens permet la recherche des signes histologiques caractéristiques d’empéripolèse et l’expression par la PS100. Les images d’empéripolèse sont d’ailleurs moins évidentes au niveau cutané [10]. La présentation tumorale chez notre malade avait également fait évoqué le diagnostic erroné de lymphome cutané qui constituait effectivement un diagnostic différentiel cliniquement. L’évolution de la MRD, en général bénigne, peut cependant être exceptionnellement fatale en cas de localisation compressive d’organes vitaux [11]. Le traitement de cette affection rare et polymorphe est mal codifié. On a proposé une abstention en l’absence de gêne fonctionnelle et un traitement systémique (chimiothérapie, corticothérapie, chirurgie, radiothérapie) en cas d’atteinte viscérale symptomatique avec des résultats variables [12,13]. La résistance au protocole CHOP chez notre malade a été déjà rapportée [14].
CONCLUSION
Nous avons rapporté l’observation d’une maladie de Rosaï-Dorfman remarquable par sa présentation cutanée tumorale et profuse, ainsi que la couleur franchement jaunâtre, pseudo-xanthomateuse et par sa résistante au protocole CHOP.
RÉFÉRENCES
1. Foucar E, Rosai J, Dorfman R. Sinus histiocytosis with massive lymphadenopathy (Rosai Dorfman disease):review of the entity. Sem Diag Pathol. 1990;7:19-73.
2. Rosai J, Dorfman RF. Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entity. Arch Pathol, 1969;87:63-70.
3. Vanhaecke C, Vignon-Pennamen MD. Maladie de Rosai-Dorfman. Ann Dermatol Vénéréol. 2012;139 761-4.
4. Cortet P, Chalopin JM, Besancenot JF, Knopf JF, Michiels R, Lorenzini JL, et al Maladie de Rosai:forme grave chez un adulte avec présence d’un anticorps antifacteur VIII. Ann Med Int. 1980;131:357-60.
5. Brenn T, Calonje E, Granter SR, Leonard N, Grayson W, Fletcher CD, et al. Cutaneous Rosai-Dorfman disease is a distinct clinical entity. Am J Dermatopathol. 2002;24:385-91.
6. Lu CI, Kuo TT, Wong W., Hong HS. Clinical and histopathologic spectrum of cutaneous Rosai-Dorfman disease in Taiwan. J Am Acad Dermatol, 2004;51:931-9.
7. Galicier L, Fieschi C, Meignin E, Clauvel JP, Oksenhendler E. Histiocytose sinusale de Rosai-Dorfman. Presse Med. 2007;36:1669–75.
8. Ngo H, Blanche S, Perrin A, Manach Y. Localisations O.R.L de la maladie de Rosai-Dorfman:a propos de 4 cas.Ann Otolaryngol Chir Cervicofac. 1987;104:551-6.
9. Ang P, Tan SH, Ong BH. Cutaneous Rosai-Dorfman disease presenting as pustular and acneiform lesions. J Am Acad Dermatol. 1999;41:335-7.
10. LaplaudF AL, Leroy D, Comoz F, Morice A, Paciencia M, Allabert C, et al. Maladie de Rosai-Dorfman cutanée pure évoluant depuis 19 ans. Ann Dermatol Venereol. 2007;134:843-6.
11. Laboudi A, Haouazine N, Benabdallah L, Arzouk N, Cherradi N, Rhou H, et al. Maladie de Rosai-Dorfman révélée par une insuffisance rénale:àpropos d’un cas. Nephr. 2001;22:53–6.
12. Pulsoni A, Anghel G, Falcucci P, Matera R, Pescarmona E, Ribersani M, et al. Treatment of sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease):report of a case and literature review. Am J Hematol. 2002;69:67–71.
13. Natkunam Y. In:Sinus histiocytosis with massive lymphadenopathy (Rosai-Dorfman disease):An update. ASH Education Program, 2004. 287-291.
14. Belembaogo E, Sanou S, Girinski JR, Nguemby Mbina C. Maladie de Rosai Dorfman. Médecine d’Afrique Noire. 1999;46:123-4.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.