|
Get Citation
|
|
|
Al-Kamel MA. Keratoacanthoma of the nose coexisted with xeroderma pigmentosum in a Yemeni child: A rare case. Our Dermatol Online. 2016;7(4):419-421. |
|
|
Download citation file:
|
Keratoacanthoma of the nose coexisted with xeroderma pigmentosum in a Yemeni child: A rare case
1Regional Leishmaniasis Control Center (RLCC), P.O. Box 12692, Sana’a, Yemen, 2Dermatology Department, Sana’a University Hospital, Sana’a, Yemen
ABSTRACT
Xeroderma pigmentosum (XP) is a rare, progressive, degenerative, autosomal recessive genodermatosis. Keratoacanthoma (KA) is a rapidly growing skin tumor, occurring primarily in older men. XP associated with KA has been infrequently reported. Here, We report a rare case of KA of the nose associated with XP in an 8-year-old Yemeni child. A KA in child should suggest consideration of XP. Early medical follow up can prevent developing, and help to diagnose tumors at a curable stage before they spread out.
Key words: Keratoacanthoma; Xeroderma pigmentosum; Squamous cell carcinoma; Leonine face; Yemen
INTRODUCTION
Xeroderma pigmentosum (XP) is a rare, progressive, degenerative, autosomal recessive genodermatosis. Clinical manifestations of XP include mild to extreme sensitivity to ultraviolet radiation resulting in inflammation and neoplasia in sun-exposed areas of the skin, mucous membranes, ocular surfaces and occasionally, neurologic degeneration [1].
XP has a prevalence of about 1:1,000,000 in the U.S and Europe, for example, with higher figures in some Asian and African countries, particularly in communities with a high degree of consanguinity. XP patients have a greater than 1000-fold increase in the incidence sunlight-associated skin cancer [1,2].
Keratoacanthoma (KA) is a rapidly growing skin tumor, occurring primarily in older men, but rarely in children. XP associated with KA is extremely rare in children.
We describe here the clinical features of XP with probable KA in an 8-year-old Yemeni child with a leonine face.
CASE REPORT
In February 26, 2016, an offspring of consanguineous parents, 8-year-old Yemeni tribal male was presented to office complaining of a big mass on the nose. On inspection, the child was mentally retarded, uncooperative and photosensitive. Dermatologic examination showed (Figs. 1 and 2); leonine face; an about 33 mm × 30 mm, well-circumscribed, hemispherical tumor on the right ala of his nose, with a rough surface, brown patches and telangiectasias (closely resembles Keratoacanthoma); xerosis, different-sizes hyper- and hypo-pigmented freckle-like macules and some spots of actinic keratosis, on the face and the other exposed parts of his body. On ocular examination photophobia, bilateral corneal opacity and conjunctivitis, were observed. History revealed that; pigmentary lesion started to develop at the first year after birth; tumor started to develop around 18 months ago; and a history of continuous sun exposure.
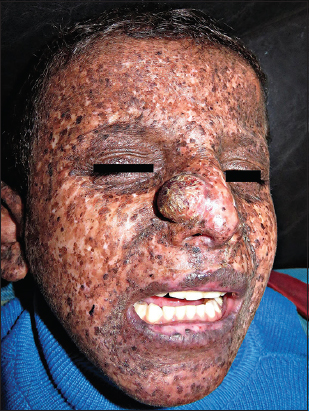
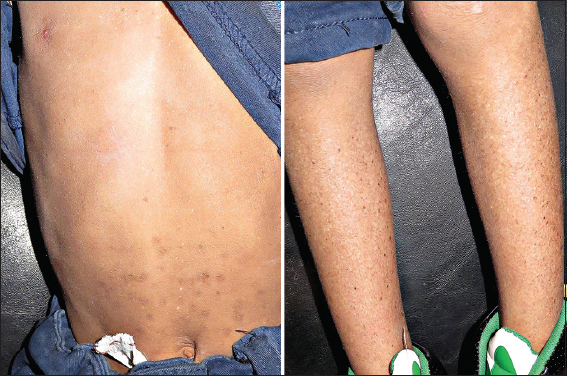

Diagnosis of Xeroderma pigmentosum (XP) associated with keratoacanthoma was made on the basis of clinical findings and family history.
On the day of presentation, tumor was excised and reported histopathology.
Patient was given supplementary vitamins, had been advised to avoid sun exposure, apply sunscreen and wear sunglasses, and then referred for other systems’ investigation.
DISCUSSION
Xeroderma pigmentosum (XP) was first described in 1874 by Hebra and Kaposi. In 1882, Kaposi coined the term “XP” for the condition referring to its characteristic dry, pigmented skin [1,2].
XP is a rare irreversible disorder, based on a genetic defect in the DNA repair system, transmitted in an autosomal recessive manner, diagnosed in early childhood and caused by exposure to ultraviolet radiation that shows that the skin is sensitive to light. It frequently manifests as extreme photosensitivity (usually the first sign in about 60% of patients), skin desquamation, freckle-like pigmentation in sun-exposed areas as early as two years of age (in 40% of patients), and may result in tumors on the skin and viscera, as well as damage to the eyes (in approximately 40% of XP patients). Therapeutic procedures for XP are based on the protection from sunlight exposure, and surgery is an effective therapeutic regimen for XP patients with complicated tumors. Prognosis is poor, and the majority of patients do not reach adult, but die from metastatic cutaneous malignancies [1–3].
Most clinical features are subject to amount of exposure to sunlight, the complementation group, the mutation, and some unknown factors. Factors aggravating the skin lesions include sunny weather, outdoor living, fair skin, smoking, poor accessibility of diagnostic amenities and improper shield from sunlight [1].
Keratoacanthoma (KA) is a rapidly growing cutaneous tumor that most commonly presents as a dome-shaped nodule with a central keratin-filled crater skin tumor, regarded as midway between a benign and malignant tumor or as a malignant tumor, equivalent to a highly differentiated squamous cell carcinoma (SCC). These tumors occur mostly on the head and face of middle-aged and elderly adults with fair complexions. There have been few reports of the associations of KA with XP. XP associated with KA is extremely rare in children. A KA in child should suggest consideration of XP [1–4].
To sum up, We reported a rare case of KA of nose coexisted with XP in a young Yemeni child.
CONCLUSION
Patients with xeroderma pigmentosum (XP) are likely to develop skin tumors like keratoacantoma (KA). Early medical follow up can prevent developing, and help to diagnose tumors at a curable stage before they spread out.
REFERENCES
1. Hasan S, Saeed S, Xeroderma Pigmentosum-A rare genodermatosis: Overview of LiteraturePigment Dis 2015; 2: 230-
2. Kleijer WJ, Laugel V, Berneburg M, Nardo T, Fawcett H, Gratchev A, Incidence of DNA repair deficiency disorders in Western Europe: Xeroderma pigmentosum, Cockayne syndrome and trichothiodystrophyDNA Repair (Amst) 2008; 7: 744-50.
3. Lehmann AR, McGibbon D, Stefanini M, XerodermapigmentosumOrphanet J Rare Dis 2011; 6: 70-
4. Gleich T, Chiticariu E, Huber M, Hohl D, Keratoacanthoma: A distinct entity?Exp Dermatol 2016; 25: 85-91.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.