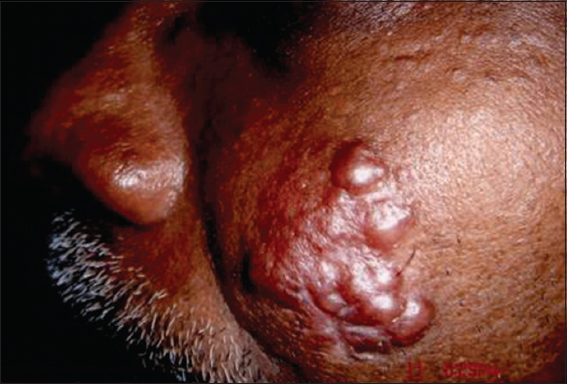
Cutaneous Rosai–Dorfmans nodules on cheek
Sundaramoorthy M. Srinivasan, Suganthi Parthasarathy
Chettinad Hospitals and Research Institute, Kelambakkam, Tamilnadu, India
ABSTRACT
Sinus histiocytosis with massive lymphadenopathy (SHML) or Rosai–Dorfman disease (RDD) is a benign idiopathic histiocytic proliferative disorder that commonly involves the lymph nodes but secondarily involves the skin. However, purely cutaneous disease without lymphadenopathy or internal organ involvement may rarely occur. We report a patient who presented with asymptomatic non-specific enlarging skin nodules without evidence of lymphadenopathy or internal disease. Histopathological examination of skin lesions in this patient showed proliferation of large histiocytes with phagocytosed inflammatory cells characteristic of Rosai–Dorfman disease. The diagnosis of purely cutaneous Rosai – Dorfman disease may be complicated by its rarity, nonspecific clinical appearance of skin lesions and broad histopathological differential diagnosis of this disorder. The prognosis is excellent in most cases. Complete spontaneous regression is known to occur. Both physicians and pathologists need to be aware of rare conditions which masquerade as lymphoproliferative disorders. Here we describe one such clinical condition – “Cutaneous Rosai Dorfman disease” and an approach to such patients.
Key words: Rosai – Dorfman disease; Sinus histiocytosis; Massive lymphadenopathy; Emperipolesis
INTRODUCTION
Most cases of Rosai – Dorfman disease present with painless cervical lymphadenopathy. The disease can involve both nodal and extranodal sites, including the skin. It is generally considered a benign, self limited proliferation of histiocytes. Although cutaneous involvement in RDD is common, purely cutaneous disease is rare. It may present at any age and is common in the second decade. Afro – Caribbeans are frequently affected.
Two etiological hypothesis have been proposed. One favours disturbance in cell mediated immunity and the other proposes the role of infection due to Epstein – Barr virus, Klebsiella, Brucella or human herpesvirus 6 as the causative agent [1].
CASE REPORT
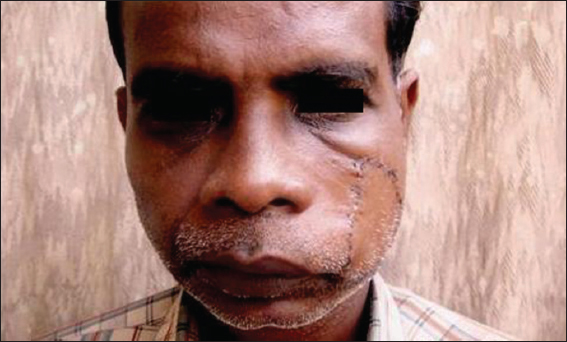
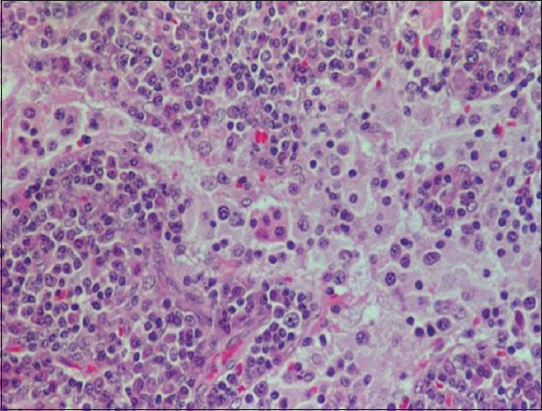
An otherwise healthy 55 year old man presented with a 2 years history of nonpruritic lesions, slowly growing as multinodular skin coloured non inflammatory nodules on the left cheek (Fig. 1). The lesion was excised by a surgeon (Fig. 2). After 6 months of postoperative period, he started developing new nodules over the site of excision. It was totally asymptomatic. The patient had no lymphadenopathy or hepatospleenomegaly. Biopsy specimens from lesions showed nodular dermal aggregates of foamy histiocytes among neutrophils, lymphocytes and plasma cells. The histiocytic nuclei were uniform, round and vesicular. Phagocytosed plasma cells and lymphocytes were seen with abundant eosinophils. The histopathology report confirmed Rosai- Dorfman disease. Histopathology was very characteristic showing dermal infiltrate composed predominantly of histiocytes with large vesicular nuclei, abundant pale cytoplasm and emperipolesis of histiocytes (Fig. 3). Typically the characteristic histiocytes of RDD were positive for S -100, negative for CD1a and variably positive for CD68. Total excision was done by a plastic surgeon with total reconstruction of the excision site. Postoperative period was uneventful. After one year, patient showed no signs of recurrence as in our picture below.



Prior to the study, patient gave written consent to the examination and biopsy after having been informed about the procedure.
DISCUSSION
The term Cutaneous Rosai – Dorfman disease is used mostly for the forms of the disease in which involvement is restricted only to the skin, in order to differentiate it from sinus histiocytosis with massive lymphadenopathy in which there is systemic involvement of multiple sites including the skin. Extranodal forms occurs in about 43% of cases, out of which skin is the most common site [2]. The etiology of the disease still remains unclear, pointing towards a reactive process against infectious agents.
The purely cutaneous forms of the disease is more common in the older age groups when compared to the systemic forms. The most common site of lesions in CRDD is the face, followed by the back, chest, thigh, flank and shoulder. The lesions may resemble psoriasis or acne [3]. CRDD shows a marked female predominance (2:1). The majority of RDD patients are of African descent and the disease is rarely reported in Asian patients. Systemic RDD is more prevalent, while the pure cutaneous form accounts for only 3% of RDD [4] and involves only the skin and adjacent soft tissues without associated lymphadenopathy. Purely CRDD was reported for the first time by Thawerani et al in 1978 in a 48 year old male patient who presented with a solitary nodule on the shoulder [5]. Patients with CRDD are generally of normal health without fever, malaise, night sweat or other associated immune deficiency symptoms. Skin lesions are divided into three main types as papulonodular, indurated plaque and tumour. Based on literature, approximately 10% of patients have had cutaneous infiltrates, but very few of them have had lesions limited to the skin [6]. Cutaneous manifestations develop in about 10% of patients, characterized by asymptomatic xanthoma like, yellowish or reddish brown papules, nodules, and plaques which may ulcerate [7].
The retrospective review of the medical literature shows the following statistics in Tables 1 and 2.


Histopathology is very characteristic showing dermal infiltrate composed predominantly of histiocytes with large vesicular nuclei, abundant pale cytoplasm and emperipolesis of histiocytes. Emperipolesis means lymphocytophagocytosis (intact lymphocytes within the cells) which differs from phagocytosis in that lymphocytes taken up are not attacked by enzymes and appear intact within the histiocytes. Emperipolesis, a consistent finding in nodal disease, is often less apparent in extranodal sites and could be confidently identified in only 7 of 11 cases by hematoxylin and eosin staining morphology. Cutaneous sinus histiocytosis can be specifically diagnosed by skin biopsy [8].
The distinctive histiocytes of RDD exhibit emperipolesis, association with numerous plasma cells and distention of lymph node sinuses or lymphatics in extra nodal sites. This helps in differentiating RDD from a variety of benign and malignant disorders such as melanoma and cancer metastasis to lymph nodes, in which phagocytosis of cells may be a prominent feature. Given the wide range of clinical presentations and the broad pathological differential diagnosis, the clinical hallmark of massive lymphadenopathy is often crucial for diagnosis of SHML. Immunophenotyping studies suggest that RDD can affect the antigen presenting activity of the skin dermal dendrocyte [9]. Although pathological evaluation is a key to definitive diagnosis, the variable presence of fibrosis, vascular proliferation, neutrophil microabscesses, lymphoid aggregates with germinal centers and background histiocytic proliferations of foam cells, multinucleated giant cell/or Touton cells may result in confusion of this disorder with a neoplastic, xanthomatous, infectious or other histiocytic process [10].
Clinical course of RDD can vary with some lesions healing spontaneously and others persisting for years or recurring after excision. In cases where it progresses to affect other organs, autoimmune hemolytic anemia, lymphocytopenia and neutropenia are common findings.
The differential diagnosis of RDD includes lymphoma, malignant histiocytosis, disseminated tuberculosis, and Langerhans cell histiocytosis (LCH). The phenomenon of emperipolesis is central in differentiating RDD as the rest of these diseases fail to exhibit lymphophagocytosis. Presence of weight loss, night sweats, hepatospleenomegaly and malignant cells staining positive for CD45 favours the diagnosis of lymphoma. Malignant histiocytosis differs from RDD clinically by its rapid downhill course and pathologically by the presence of malignant histiocytes having bizarre, pleomorphic nuclei. The histiocytes in LCH have a characteristic folded and grooved nucleus and exhibit CD1a positivity. Disseminated tuberculosis can be ruled out on the basis of absence of granulomas and negative staining for acid fast bacilli by Ziehl-Neelsen stain. The presence of characteristic histiocyte derived from circulating mononuclear cells, long history and an increased incidence of serum autoreactive antibodies during active disease suggest a possible pathogenic correlations with a dysregulatory process.
It is conceivable that some patients with purely cutaneous symptoms may have clinically undetectable systemic lesions. Lesions can mimic panniculitis [11]. However there have been no reported cases of cutaneous disease developing into systemic disease. The clinical and histopathological course is variable with some lesions resolving spontaneously over weeks to months. Histopathology is very characteristic showing emperipolesis of histiocytes [12]. Few lesions can also mimic vasculitis [13,14]. Pustular lesions may appear as acneiform eruptions and others persisting for years [15] or even recurring after excision as in this reported case. Few patients can also present as granuloma annulare, which is not associated with adenopathy. Treatment is generally not necessary for cutaneous RDD, but may be desired for cosmetic purposes or symptomatic relief. Cutaneous lesions have been reported to respond to radiotherapy, cryotherapy, excision, oral and topical corticosteroids and high dose thalidomide. Dapsone and thalidomide have been effective in cases refractory to other treatments. Steroid responsive CRDD can be associated with bilateral anterior uveitis and hypothyroidism. Many cases have reported improvement with combined application of Compound betamethasone and lidocaine, and intramuscular interferon and oral acitretin. But this mechanism requires further studies in the future. Utikal et al described a patient with complete remission of CRDD after receiving imatinib therapy, however a different study reported a patient with CRDD who was completely resistant to this treatment. Investigation of the true efficacy of any of these therapeutic modalities is complicated by the rarity of this disorder.
Scrutiny of the dermatological literature yields few cases of malignant histiocytosis presenting in the skin and breast using restrictive criteria as defined by Pileri et al (a malignant neoplasm of histiocytes, positive for one or more histiocytic markers but negative for accessory/dendritic cell markers. Several cases in the past have also been diagnosed as such, but do not adhere to the latest criteria/or lack the appropriate immunostaining techniques. To the best of our knowledge, this is the first case reported that adheres to Pileri ‘s criteria, yet does not show any evidence of systemic involvement.
Marshall in 1981 reviewed 320 cases based on literature, of which 6.9% presented chiefly with cutaneous lesions. These have been described on the face, scalp, chest, back and abdomen and more frequently on the extremities, especially the legs. Men are more often affected than women (2:2:1). Laboratory investigations may reveal pancytopenia, a raised ESR, eosinophilia, leukocytosis, altered liver function tests particularly increased aspartate aminotransferase, prolonged prothrombin time and cholestasis(in the absence of drug abuse or alcohol history).
CONCLUSION
Cutaneous Rosai Dorfman disease is a very rare cutaneous histiocytosis. When associated with lymphadenopathy and hepato-spleenomegaly, it can be fitted with sinus histiocytosis and the diagnosis is easier. Though we read about C-RRD, practical application of our knowledge is very meagre. This case report and literature survey emphatically emphasizes that histopathology is the concluding material to all dermatologists.
REFERENCES
1. Rosai J, Dorfman RF, Sinus histiocytosis with massive lymphadenopathy. A newly recognized benign clinicopathological entityArch Pathol 1969; 87: 63-70.
2. Pitamber HV, Grayson W, Five cases of cutaneous Rosai Dorfman diseaseClin Exp Dermatol 2003; 28: 17-21.
3. Uniyal SK, Beena KR, Cutaneous Rosai – Dorfman disease preceeding inguinal lymphadenopathyInt J Dermatokl 2002; 41: 404-6.
4. Van Zander J, Cutaneous Rosai – Dorfman diseaseDermatol Online J 2004; 10: 12-
5. Thawerani H, Sanchez RL, Rosai J, Dorfan RF, The cutaneous manifestations of sinus histiocytosis with massive lymphadenopathyArch Dermatol 1978; 114: 191-7.
6. Lazar AP, Esterly NB, Gonzalez-Crussi F, Sinus histiocytosis clinically limited to the skinPediatr Dermatol 1987; 4: 247-53.
7. Lu C, Kuo T, Wong W, Hong H, Clinical and histopathologic spectrum of cutaneous Rosai – Dorfman disease in TaiwanJ Am Acad Dermatol 2004; 51: 931-9.
8. Chu P, Le Boit PE, Histologic features of cutaneous sinus histiocytosis (Rosai-Dorfman disease): study of cases both with and without systemic involvementJ Cutan Pathol 1992; 19: 201-6.
9. Perrin C, Michiels JF, Lacour JP, Chagnon A, Fuzibet JG, Sinus histiocytosis (Rosai – Dorfman disease) clinically limited to the skin. An immunohistochemical and ultrastructural studyJ Cutan Pathol 1993; 20: 368-74.
10. Quaglino P, Tomasini C, Novelli M, Colonna S, Berneno MG, Immunohistologic findings and adhesion molecule pattern in primary pure cutaneous Rosai – Dorfman disease with xanthomatous featuresAm J Dermatopathol 1998; 20: 393-8.
11. Puppin D, JrChavaz P, Harms M, Histiocytic lymphophagocytic panniculitis (Rosai – Dorfman disease): a case reportDermatology 1992; 184: 317-20.
12. Skiljo M, Garcia Lora E, Tercedor J, Massare E, Esquivias J, Garcia–Mellado V, Purely cutaneous Rosai – Dorfman diseaseDermatology 1995; 191: 49-51.
13. Suster S, Cartagena N, Cabello–Inchausti B, Robinson MJ, Histiocytic lymphophagocytic panniculitis. An unusual extranodal presentation of sinus histiocytosis with massive lymphadenopathy (Rosai – Dorfman disease)Arch Dermatol 1988; 124: 1246-9.
14. Stefanato CM, Ellerin PS, Bhawan J, Cutaneous sinus histiocytosis (Rosai Dorfman disease) presenting clinically as vasculitisJ Am Acad Dermatol 2002; 46: 775-8.
15. Ang P, Tan SH, Ong BH, Cutaneous Rosai – Dorfman disease presenting as pustular and acneiform lesionsJ Am Acad Dermatol 1999; 41: 335-7.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.