Phakomatosis pigmentovascularis with lower limb vascular abnormalities in a young Kashmiri male child-Report of a first child from Kashmir Valley (India) and review of literature
Majid Jehangir1, Seema Quyoom2, Jahangeer Bhat1, Peerzada Sajad2, Ishfaq Sofi3, Aresalan Amin1, Mudasir Bhat1
1Department of Radiodiagnosis and Imaging, Government Medical College, Srinagar (University of Kashmir), J&K, India, 2Department of Dermatology, SKIMS Medical College Srinagar, Bemina, Srinagar, Kashmir, J&K, India[3] Postgraduate, Department of Ophthalmology, Government Medical College, Srinagar (University of Kashmir), J&K, India
ABSTRACT
Phakomatosis is a developmental abnormality simultaneously involving eyes, central nervous system, and skin. Phakomatosis pigmentovascularis (PPV) is a rare cutaneous disorder which is characterised by a combination of capillary malformations and pigmented anomalies. It arises sporadically. PPV was first described by Ota et al., in 1947. There is no sex predilection, but Japanese have been found to be affected more. There are four main types of PPV. Recently a fifth type with cutis marmorata and aberrant Mongolian blue spot has also been added to the classification. Here we report a case of PPV with Struge – Weber syndrome and Klippel Trenaunay syndrome in a young Kashmiri male child, which has been rarely reported in the literature.
Key words: Phakomatosis pigmentovascularis(PPV); Klippel Trenaunay syndrome(KTS); Struge – Weber syndrome(SWS)
INTRODUCTION
Phakomatosis, a neurocutaneous syndrome is a developmental abnormality which is characterised by the involvement of skin, eyes and central nervous system. Phakomatosis pigmentovascularis is a rare cutaneous disorder which was first described by Ota et al. in 1947 [1]. There is no sex predilection. Four types with two subtypes have been described, where subtype ‘a’ refers to cases presenting with only cutaneous manifestations and subtype ‘b’ is characterised by systemic association like in, Struge – Weber syndrome and Klippel Trenaunay syndrome [2]. Recently a fifth type with cutis marmorata and aberrant Mongolian blue spot has also been added to the classification.
CASE REPORT
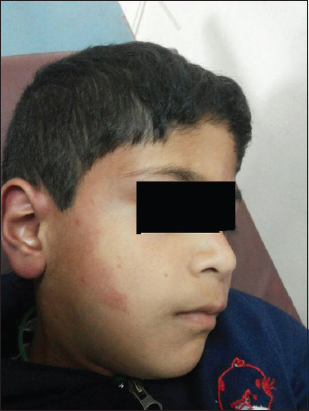
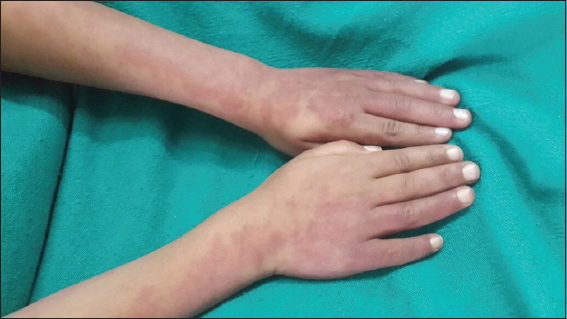
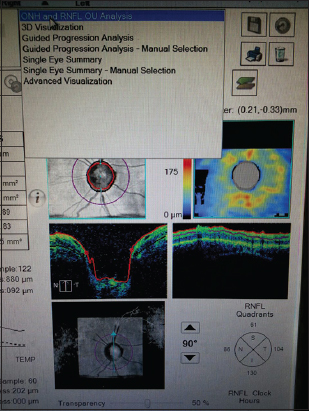
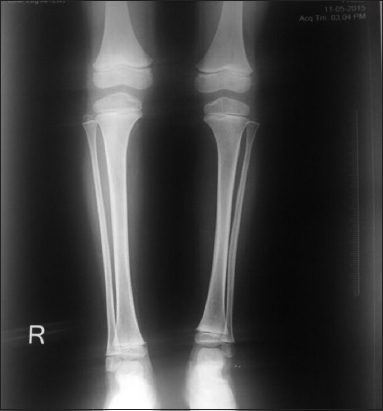
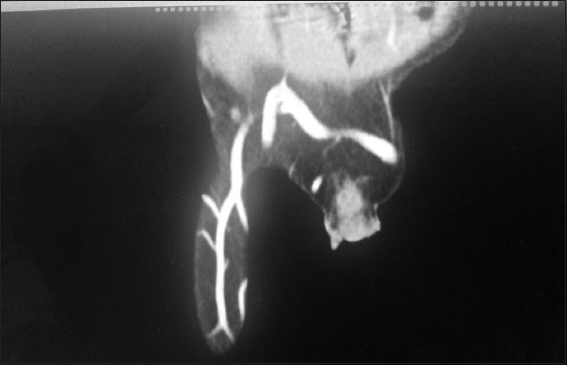
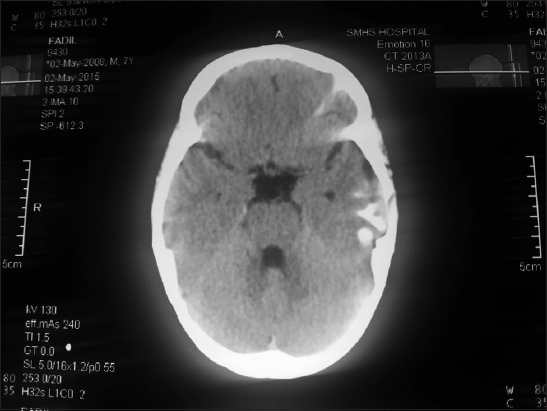
A 12 – year old male child product of a non-consanguinous marriage presented with history of non-blanchable erythematous macules, which was noted to be portwine stain (naevus flammeus) on examination involving face, forearms, and right lower limb since birth (Fig. 1 and 2). There was history of increase in the size of these macules with age. The facial portwine stain was more marked and abundant on the left side. There was associated history of abnormal gait. However there was no history of headache, seizures, abnormal body movements or blurring of vision. The patient was noted to have hypertrophy of right lower limb with disparity in the length of legs. A visible vessel was seen in right groin crossing the midline (Fig. 3). Ophthalmological examination revealed raised intraocular pressure in left eye. Optical coherence tomography revealed severe thinning of Retinal nerve fibre layer (RNFL) in left eye, loss of neuro-retinal rim (NRR), gross asymmetry in cup-disc ratio and RNFL thickness (Fig. 4 and 5). X ray of lower limbs showed limb lengthing in right limb, however, no bony hypertrophy was seen (Fig. 6). Colour Doppler and CT angiography of limbs revealed absence of right iliac vein with right femoral vein draining through collateral vein crossing midline into left external iliac vein. High origin of superficial femoral artery and deep femoral artery was also noted (Fig. 7). Non contrast CT of brain showed foci of dense gyral calcification in left temporoparietal region (Fig. 8).


DISCUSSION
The term phakomatosis refers to a developmental abnormality simultaneously involving skin, eyes and central nervous system. Phakomatosis pigmentovascularis is a rare syndrome characterised by the combination of a capillary malformation with pigmented naevi of various types. It is very uncommon and arises sporadically. Both, males and females are affected. There is no sex predilection, but Japanese have been reported to be affected more often [1,2]. Twin spotting phenomenon of Happle and Steijen has been proposed to explain this phenomenon (PPV). In this hypothesis, somatic mutations on nearby genes leads to mosaic spots in close proximity to one another [3]. PPV has been classified into four types namely; type I: Nevus flammeus and epidermal nevus, type II: Nevus flammeus, Mongolian spots, ± nevus anemicus, type III: Nevus flammeus, nevus spilus, ± nevus anemicus, and type IV: Nevus flammeus, Mongolian spots, nevus spilus, ± nevus anemicus. Recently a fifth type with cutis marmorata and aberrant Mongolian blue spot has also been added to the classification. Each type is subdivided into two subtypes; subtype ‘a’, cutaneous involvement only; and subtype ‘b’, both cutaneous and systemic involvement. Type II is reported to account for approximately 80% of all cases. PPV type I, III, and IV have very rarely been reported in literature. The capillary malformation may occur anywhere on the body and can be segmental. The aberrant Mongolian spots occur on atypical sites and tend to persist, unlike conventional Mongolian blue spots. It is estimated that 50% of patients with PPV present with systemic involvement. The cases that have been most studied are those associated with PPV type IIb. In the case of PPVs, neurologic anomalies develop in the first months of life. The most common ocular abnormality which has been found to be associated with PPV is ocular melanosis, which consists of unilateral or bilateral blue-gray pigmentation in the sclera (Naevus of Ota). Other ophthalmologic abnormalities are much less common. However, a large number of abnormalities have been published in relation to PPVs, but with the exception of Sturge-Weber syndrome, Klippel-Trenaunay syndrome, and ocular melanosis, the abnormalities are varied [4,5].
It is recommended that all the affected cases should be investigated depending on the sites involved, like for glaucoma if the capillary malformation affects the eyelids, or for Klippel-Trenaunay syndrome if there is limb involvement. A patient’s quality of life may be improved by treating nevus flammeus using a pulsed dye laser and by treating pigmentary nevus using Q-switched lasers. Some authors suggest that the pigmentary nevus should be treated first [6].
Various systemic syndromes have been reported to be associated with PPV in the literature, and these include; Sturge-Weber syndrome, Nevus of Ota, and Klippel-Trenaunay syndrome [7].
Sturge –Weber syndrome (SWS) is defined as a facial portwine stain with ipsilateral vascular malformation of leptomeninges and eyes. Eye involvement is however not necessary for the diagnosis of SWS. Brain changes show calcification with atrophy and increased vascularity of unilateral leptomeninges.
Naevus of Ota also known as naevus fuscoceruleus ophthalmomaxillaris is a dermal melanocytosis and is characterised by slate-grey or bluish hyperpigmentation involving the periorbital area, sclera and conjunctiva. It may be congenital or may develop around puberty.
Klippel Trenaunay syndrome is characteried by the association of cutaneous capillary malformation of limbs(naevus flammeus) and soft tissue swelling of limb with or without bone hypertrophy. Original description of this syndrome included vericosities of the limb [8,9].
Since its first description in 1947, 222 cases of PPV have been published, most of which are sporadic and from Japan, Mexico, or Argentina. The most common type is type IIb (45%), followed by IIa (30%). The rest are much less frequent and no isolated cases of PPV Ib, IVb, or Vb have been published. To date only two case series of PPV have been published. The largest series was published by Cordisco et al [10], who presented 25 patients in Argentina. In this series, type IIb was the most common type. Vidaurri-de la cruz et al [11] presented a series of 24 patients in Mexico in which 75% of the cases were type IIb, 25% were IIa, and the rest of the subtypes were not found.
Literature shows that till date only few well established cases of phakomatosis pigmentovascularis (PPV) have been reported, but PPV presenting with Sturge-Weber syndrome and lower limb vascular abnormalities is extremely rare. Sumita S et al [12] reported a case of PPV with Sturge–Weber syndrome and Klippel-Trenaunay Syndrome in a 13 year old female who presented with port wine stain over face, limbs and trunk, with a visible vessel along the lateral aspect of left lower leg together with associated hypertrophy of the involved limb.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Ota M, Kawamura T, Ito N, Phakomatosis pigmentovascularisDermatol Surg 1947; 52: 1-3.
2. Marti-Bonmati L, Menor F, Mulas F, The Struge – Weber syndrome: correlation between the clinical status and radiological CT AND MRI findingsChilds Nervs Syst 1993; 9: 107-9.
3. Danarti R, Happle R, Paradominant inheritance of twin spotting: phakomatosis pigmentovascularis as a further possible exampleEur J Dermatol 2003; 13: 612.
4. Young AE, Mulliken JB, Young AE, Combined vascular malformationsVascular birthmarks: Haemangiomas and malformations 1988; Philadelphia: WB Saunders; 246-74.
5. Happle R, Stejlen PM, Phacomatosis pigmentovascularis interpreted as a phenomenon of twin spotsHautarzt 1989; 40: 721-24.
6. Du LC, Delaporte E, Catteau B, Destee A, Piette F, Phakomatosis pigmentovascularis type IIEur J Dermatol 1998; 8: 569-72.
7. Gupta A, Dubey S, Agarwal M, A case of Sturge-Weber syndrome in association with phacomatosis pigmentovascularis and developmental glaucomaJ AAPOS 2007; 11: 398-9.
8. Goyal T, Varshney A, Phacomatosis cesioflammea: First case report from IndiaIndian J Dermatol Venereol Leprol 2010; 76: 307.
9. Sawada Y, Iwata M, Mitsuhashi Y, Nevus pigmentovascularisAnn Plast Surg 1990; 25: 142-5.
10. Cordisco MR, Campo A, Castro C, Bottegal F, Bocian M, Persico S, Phakomatosis pigmentovascularis: report of 25 casesPediatr Dermatol 2001; 18: 70.
11. Vidaurri-de la cruz H, Tamayo-Sanchez L, Duran-McKinster C, Orozco-Covarrubias ML, Ruiz-Maldonado R, Phakomatosis pigmentovascularis IIa and IIb: clinical findings in 24 patientsJ Dermatol 2003; 30: 381-8.
12. Sumita S, Sanchaita B, Chinmay H, Rahul A, Anusree G, Phakomatosis pigmentovascularis presenting with Sturge-Weber syndrome and Klippel-Trenaunay syndromeIndian J Dermatol 2015; 60: 77-81.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.