A Moroccan case of piebaldism in an infant as part of a family phenotype
Mohamed El Amraoui , Meryem Khalidi, Rachid Frikh, Naoufal Hjira, Mohammed Boui
, Meryem Khalidi, Rachid Frikh, Naoufal Hjira, Mohammed Boui
Department of Dermatology, Mohammed V Military Training Hospital, Rabat, Morocco
Corresponding author: Dr. Mohamed El Amraoui
Submission: 06.10.2019; Acceptance: 13.01.2020
DOI: 10.7241/ourd.2020e.178
Cite this article: El Amraoui M, Khalidi M, Frikh R, Hjira N, Boui M. A Moroccan case of piebaldism in an infant as part of a family phenotype. Our Dermatol Online. 2020;11(e):e178.1-e178.2.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
Sir,
Hypopigmentation in children is always a reason for particular consultation with a considerable emotional burden of parents whose main obsession is not to have a vitiligo, however other disorders of melano genesis often hereditary and sometimes syndromic may be prognostic more reserved. Piebaldism is in the intermediate zone.
Female infant, 105 days old, from a non-consanguineous marriage. Was presented for dermatological consultation by her mother, for achromic macules in the body, roughly bilateral and symmetrical, with a triangular hypo chromic macula on the forehead and a white wick of the scalp appeared from birth (Figs. 1a and 1b). The mother reports the existence of a white wick in the two sisters, in herself, in the maternal grandfather and in other maternal descendants.The neurological, ophthalmological and ENT examinations were without abnormalities.The diagnosis of a piebaldism in the context of a family phenotype was retained and a genetic study was planned but which was not done for lack of means.The girl was put on topical Tacrolimus with eviction of the Koebner phenomenon.
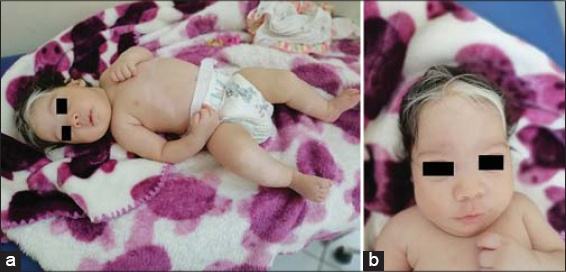 |
Figure 1:(a and b) Diffuse hypochromic and achromic macules with a frontal white wick in a child with piebaldism with similar cases in the family. |
Piebaldism is a rare genetic disorder of autosomal dominant inheritance, which is due to one of the c-Kit gene mutations (more than 60 mutations described) that are implicated in melanocytic proliferation and migration during embryogenesis. Clinically, it is manifested by achromic or hypo-pigmented macules, well circumscribed, present at birth, stable and symmetrically distributed, associated with a white wick, often of triangular shape, present on the frontal part of the scalp with sometimes Achieving the underlying forehead, however, 80-90% of patients present white wick only. Extra-skin manifestations are usually rare (hearing and vision problems). The most commonly described combinations are: type I neurofibromatosis, congenital megacolon, Blackfan-Diamond anemia, and type 1a glycogenosis. The main differential diagnoses remain albinism and Waardenburg syndrome. Diagnosis remains clinical after elimination of other differential diagnoses. Confirmation by genetic study can be considered especially in the context of genetic counseling. The treatment is generally disappointing with a significant social and psychological impact [1–3].
Piebaldism is an inherited disorder of pigmentation that is rare and has a favorable prognosis in general.
Consent
The examination of the patient wasconductedaccording to the Declaration of Helsinki principles.
The authorscertifythatthey have obtained all appropriate patient consent forms. In the form the patient(s) has/have givenhis/her/their consent for his/her/their images and otherclinical information to bereported in the journal. The patients understandthattheirnames and initialswill not bepublished and due efforts willbe made to concealtheiridentity, but anonymitycannotbeguaranteed.
REFERENCES
1. El Kouarty H, BenjellounDakhama BS. Piebaldism:a pigmentary anomaly to recognize:about a case and review of the literature. Pan Afr Med J.2016;25:155.
2. Debbarh FZ, Mernissi FZ.Piebaldisme:une génodermatose rare. Pan Afr Med J. 2017;27:221.
3. Ortonne JP. Piebaldism, Waardenburg’ssyndrome, and relateddisorders:“Neural Crest Depigmentation Syndromes“?.Dermatol Clin. 1988;6:205-16.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0001-7687-0158 http://orcid.org/0000-0001-7687-0158 |

Comments are closed.