A rare classic case of Dowling-Degos disease with dermoscopy description
Selma El Kadiri , Hanane Bay Bay, Rhizlane Chaoui, Zakia Douhi, Sara Elloudi, Fatima Zahra Mernissi
, Hanane Bay Bay, Rhizlane Chaoui, Zakia Douhi, Sara Elloudi, Fatima Zahra Mernissi
Department of Dermatology and Venerology, University Hospital Hassan II, Fez, Morocco
Corresponding author: Dr Selma El Kadiri
Submission: 11.05.2020; Acceptance: 01.07.2020
DOI: 10.7241/ourd.2020e.71
Cite this article: El Kadiri S, Bay Bay H, Chaoui R, Douhi Z, Elloudi S, Mernissi FZ. A rare classic case of Dowling-Degos disease with dermoscopy description. Our Dermatol Online. 2020;11(e):e71.1-e71.2.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
Sir,
Dowling-Degos disease (DDD) is a rare autosomal dominant disorder, classically characterized by acquired reticular hyperpigmentation starting in flexural areas [1]. Herein, we report a rare classic case of DDD with dermoscopic description.
A 42-year-old female patient presented with asymptomatic pigmented lesions started initially in the axillar area. They grew progressively to involve the full body. She had a family history of similar pigmentary disorders in her mother and a personal history of epidermoid cysts. Physical examination flat hyperpigmented macules over neck, trunk, and limbs in a reticulate pattern (Figs. 1a and b). Hypopigmented macules with surrounding hyperpigmentation were present over the limbs (Fig. 2). Face, extremities, oral mucosa, hair, and nail were spared. Dermoscopy revealed brownish irregular pigment surrounding the hypopigmented center with a reticular distribution (Fig. 3). Histologic examination of biopsy specimens revealed melanin in the epidermis, dermal melanophages, and perivascular lymphohistiocytic infiltrate compatible with the diagnosis of DDD.
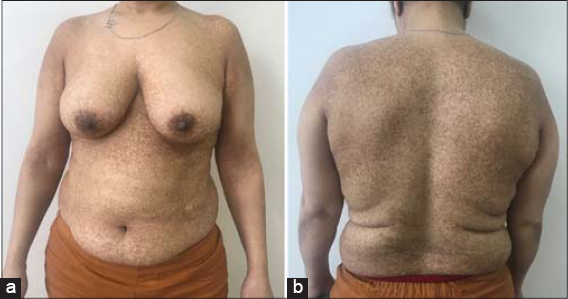 |
Figure 1: (a and b) Clincial image showing hyperpigmented macules over neck, trunk and arms. |
 |
Figure 2: Hypopigmented macules surrounded by hyperpigmented on the thighs. |
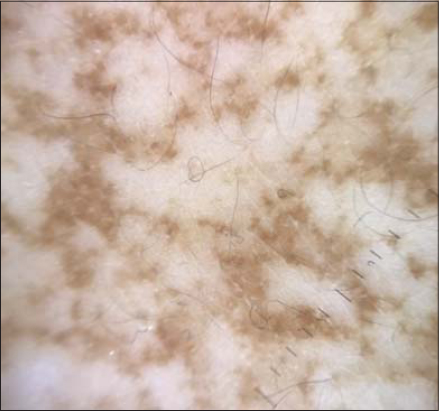 |
Figure 3: Dermoscopy showing brownish irregular pigment surrounding the hypopigmented center with a reticular distribution. |
DDD occurs initially in the third to fourth decade of life [1]. The hyperpigmentation is composed of lentigo-like brown macules with inconstant brown hyperkeratotic papules in the axillae. The pigmentation extends to the rest of the body to involve inframammary folds, trunk, arms, and thighs. Flexural areas may be pruriginous. Comedone-like lesions in the back and neck, pitted perioral scans, epidermoid cysts, and hidradenitis suppurativa have been reported in some patients [2]. DDD is caused by a loss-of-function mutation in the non-helical head domain of the keratin 5 gene [3]. Histologically, there is an increase in melanin in the basal cell layer, elongation of rete ridges, dermal melanophages, and perivascular lymphohistiocytic infiltrate [4]. The differential diagnosis includes acanthosis nigricans when only the intertriginous areas are involved but the lesions are papillomatous .and velvety. Freckles in neurofibromatosis type 1 involving axillae and groin but other features make the diagnosis easy. Haber syndrome is characterized as well with hyperpigmentation on the trunk and proximal limbs and photosensitive rosacea like facial lesions during adolescence. If the patient complains of pruritus with reticulated hyperpigmentation, prurigo pigmentosa has to be suspected and a diagnosis is made with a skin biopsy revealing lymphocytic infiltrate with multiple necrotic keratinocytesr [4]. There are only a few descriptions of dermoscopy of DDD revealing irregular brownish projections around the hypopigmented center. The irregular brownish projections correspond to the elongated rete ridges with pigmentation at tips surrounding a whitish center related to the follicular plugs [1].
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
REFERENCES
1. Nirmal B, Dongre AM, Khopkar US. Dermatoscopic Features of Hyper and Hypopigmented Lesions of Dowling Degos Disease. Indian J Dermatol. 2016;61:125.
2. Mahajan SH, Mahajan SA, Khopkar US, Kharkar VD. Follicular Dowling-Degos Disease:A Rare Pigmentary Dermatosis. Indian Dermatol Online J. 2017;8:487-9.
3. Bachaspatimayum R, Das P, Bhattacharjee N. Dyschromatosis universalis hereditaria:A rare case report from Northeast India. Our Dermatol Online. 2019;10:179-80.
4. Tod BM, Steenkamp I, Jordaan HF, Visser WI. Dowling-Degos disease presenting primarily with comedones and atrophic scarring. Dermatopathology (Basel). 2019;6:153-6.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/000-0003-3455-3810 http://orcid.org/000-0003-3455-3810 |

Comments are closed.