X-linked Ichthyosis: Image article
Alaoui Aicha Elharrouni 1, Mariame Elimam2, Chayemae Jroundi1, Sara Elloudi1, Zakia Douhi1, Hanane Baybay1, Mostapha Hida2, Fatima Zahra Mernissi1
1, Mariame Elimam2, Chayemae Jroundi1, Sara Elloudi1, Zakia Douhi1, Hanane Baybay1, Mostapha Hida2, Fatima Zahra Mernissi1
1Departement of Dermatolgy,CHU Hassan II, Fez, Morocco; 2Departement of Pediatrics,CHU Hassan II, Fez, Morocco
Corresponding author: Dr. Alaoui Aicha Elharrouni, E-mail: ealaouiaicha@gmail.com
Submission: 25.10.2019; Acceptance: 10.01.2020
DOI: 10.7241/ourd.2020e.16
Cite this article: Elharrouni AA, Elimam M, Jroundi C, Elloudi S, Douhi Z, Baybay H, Hida M, Mernissi FZ. X-linked Ichthyosis: Image article. Our Dermatol Online. 2020;11(e):e16.1-e16.2
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
Sir,
We report a case of 2 months old infant from a consanguineous marriage,presented of adherent translucent scales begin to appear in a generalized distribution and desquamate widely. A number of other families with similar clinical findings and pedigrees were reported. Clinical examination revealedpolygonal, loosely adherent translucent scales with sparing of flexures (Figs. 1a and 1b). Biologicsexaminations were normal.Patient was diagnosed as X-linked ichthyosis based on clinical features, history other families with similar clinical findings.He was prescribed topical emollients, he showed improvement in scaling after six weeks (Figs. 1c and 1d).
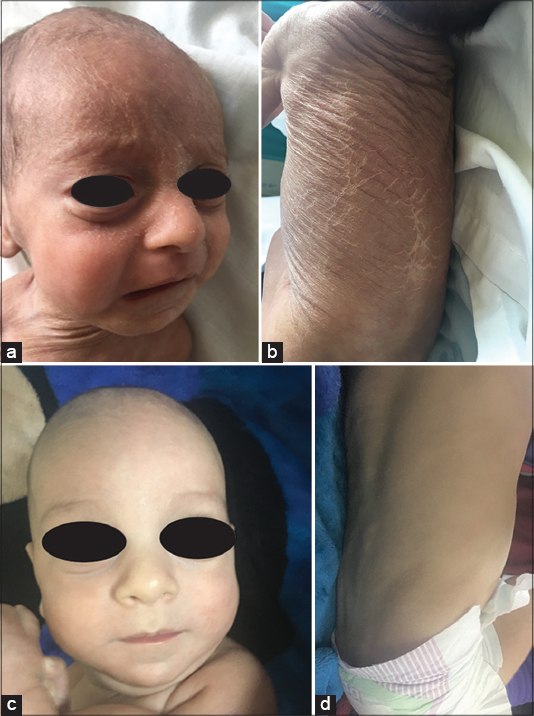 |
Figure 1. (a-d) 2 month infant presents with polygonal, loosely adherent translucent scales with sparing of flexures (a and b). And l’evolution: complete re-epithelialization within six weeks (c and d). |
There exists a group of rare, inherited scaly skin disorders, generally termed ichthyosis,are due to mutations in genes mostly involved in skin barrier formation [1]. They share as a common feature, thickening ot” the stratum corneum leading to a scaly appearance of the skin. There are two types of inherited ichthyoses: non-syndromic ichthyoses and ichthyosis syndromes [2]. On-syndromic ichthyoses are characterized by the phenotypic expression of the disorder being seen only in the skin, include ichthyosis vulgaris, recessive X-linked ichthyosis and other forms.Individual types of ichthyosis may be distinguished on clinical grounds [3]. Most patients have disease limited to the skin. The majority of cases improves with age and do require symptomatic treatment is performed with topical keratolytics and emollients [2,3],given the complex pathophysiology that aimed to repair and protect the skin barrier and improve quality of life of these patients.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
Copyright by Alaoui Aicha Elharrouni, et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
REFERENCES
1. Rodrigo-Nicol_as B, Bueno-Martinez E, Mart?n-Santiago A et al. Evidence of the high prevalence of neurological disorders in nonsyndromic X-linked recessive ichthyosis:a retrospective case series. Br J Dermatol 2018;179:933–39.
2. Neil F. Fernandes, MD,a Camila K. Janniger, MD,a,b and Robert A. Schwartz, MD, MPHa,b,c Newark, New Jersey:X-linked ichthyosis:An oculocutaneous genodermatosis, 2009 by the American Academy of Dermatology, Inc. doi:10.1016/j.jaad.2009.04.028
3. An oculocutaneous genodermatosis. J Am Acad Dermatol. 2010;62 (3):480-485.
Notes
Source of Support: Nil
Conflict of Interest: None declared
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/000-0003-3455-3810 http://orcid.org/000-0003-3455-3810 |

Comments are closed.