Keratitis-Ichthyosis-Deafness (KID) Syndrome, case report
Tomasz Wasyłyszyn1, Katarzyna Borowska2
1Military Institute of Medicine in Warsaw, Department of Dermatology, Szaserów 128, 04-141 Warsaw, Poland
2Department of Histology and Embryology with Experimental Cytology Unit, Medical University of Lublin, 11 Radziwiłłowska, 20–080 Lublin, Poland
Corresponding author: Prof. Katarzyna Borowska, E-mail: k_borowska@wp.pl
Submission: 01.08.2017; Acceptance: 10.08.2017
How to cite this article: Wasyłyszyn T, Borowska K. Keratitis-Ichthyosis-Deafness (KID) Syndrome, case report. Our Dermatol Online. 2017;8(3e):e3.
ABSTRACT
Keratitis-Ichthyosis-Deafness (KID) syndrome is a congenital disorder characterized by keratitis, ichthyosis, and deafness. Authors present a 10 year old by with KID syndrome. The diagnosis was based on a clinical picture, hearing evaluations and genetic research.
Key words: KID syndrome, mutations, keratoderma.
INTRODUCTION
Keratitis-Ichthyosis-Deafness (KID) syndrome is a congenital disorder characterized by keratitis, ichthyosis, and deafness. KID syndrome was first reported in 1915 by Burns as a generalized congenital keratoderma with ocular and mucosal involvement. Heterozygous missense mutations in the GJB2 gene localized on chromosome 13q11-q12 encoding a gap junction protein called connexin-26 were found to be associated with the KID syndrome. Both an autosomal dominant form and an autosomal recessive form have been described [1,2]. Moreover, most patients with KID syndrome harbor p.D50N gene [3]. The skin lesions, described as erythrokeratoderma occurs occasionally with scarring alopecia and are predominantly on the face, palms and soles, with a typical reticulated pattern. Additional features are corneal epithelial defects, including scarring and neo-vascularization that can result in progressive decline of visual acuity and may eventually lead to blindness. About 10-15% of KID syndrome patients are reported to develop squamous cell carcinoma (SCC) of the skin and oral mucosa [4,5].
CASE REPORT
Authors present a 10 year old by with KID syndrome. The diagnosis was based on a clinical picture, hearing evaluations and genetic research. Clinically patient represented ichtyosis of a very severe grade affecting practically the whole body. Unlike ichtyolis vulgaris, where joint areas are free of lesions [6], these areas were most severely affected in the present patient (Figs. 1, 3, 4). Moreover thick hyperkeratotic lesions were observed on the central parts of the face (Fig. 2), especially on the forehead, between the brows. The latter ones led to irreversible alopecia of the brows and lashes (Fig. 2). Unlike written in the literature alms ad soles were moderately affected. Patient suffered photophobia and opened his eyes only partially even in a dark room (Fig. 5).
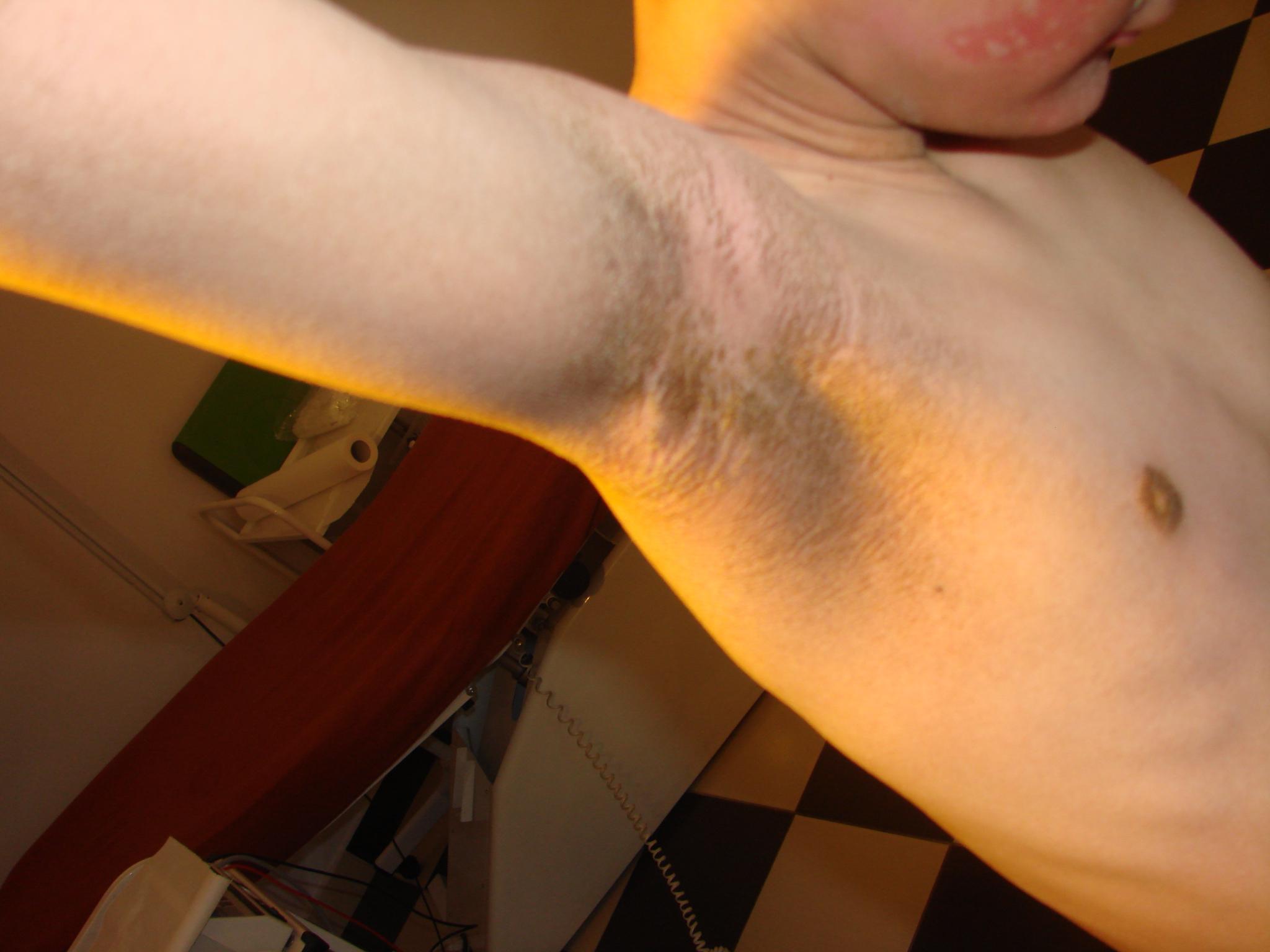

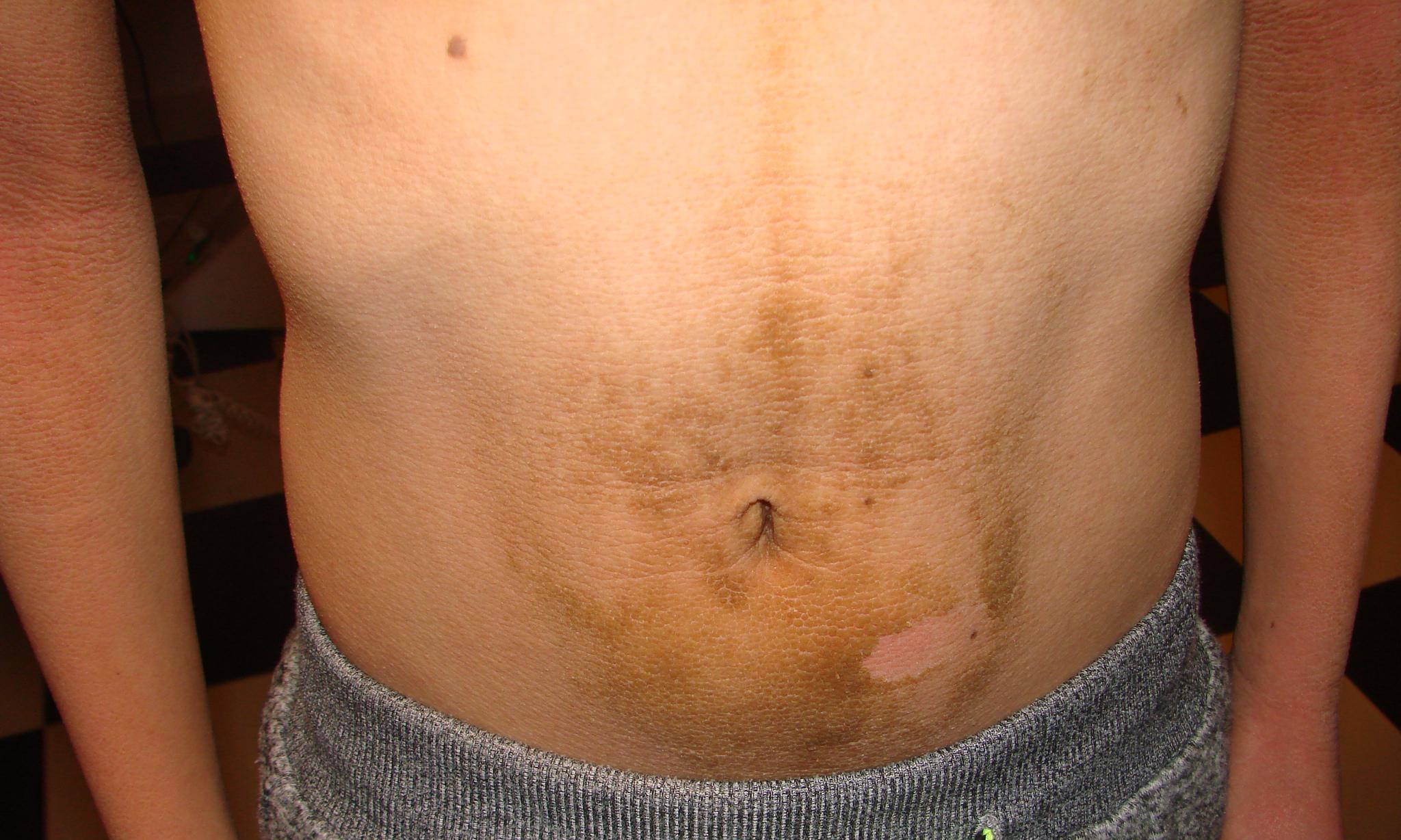

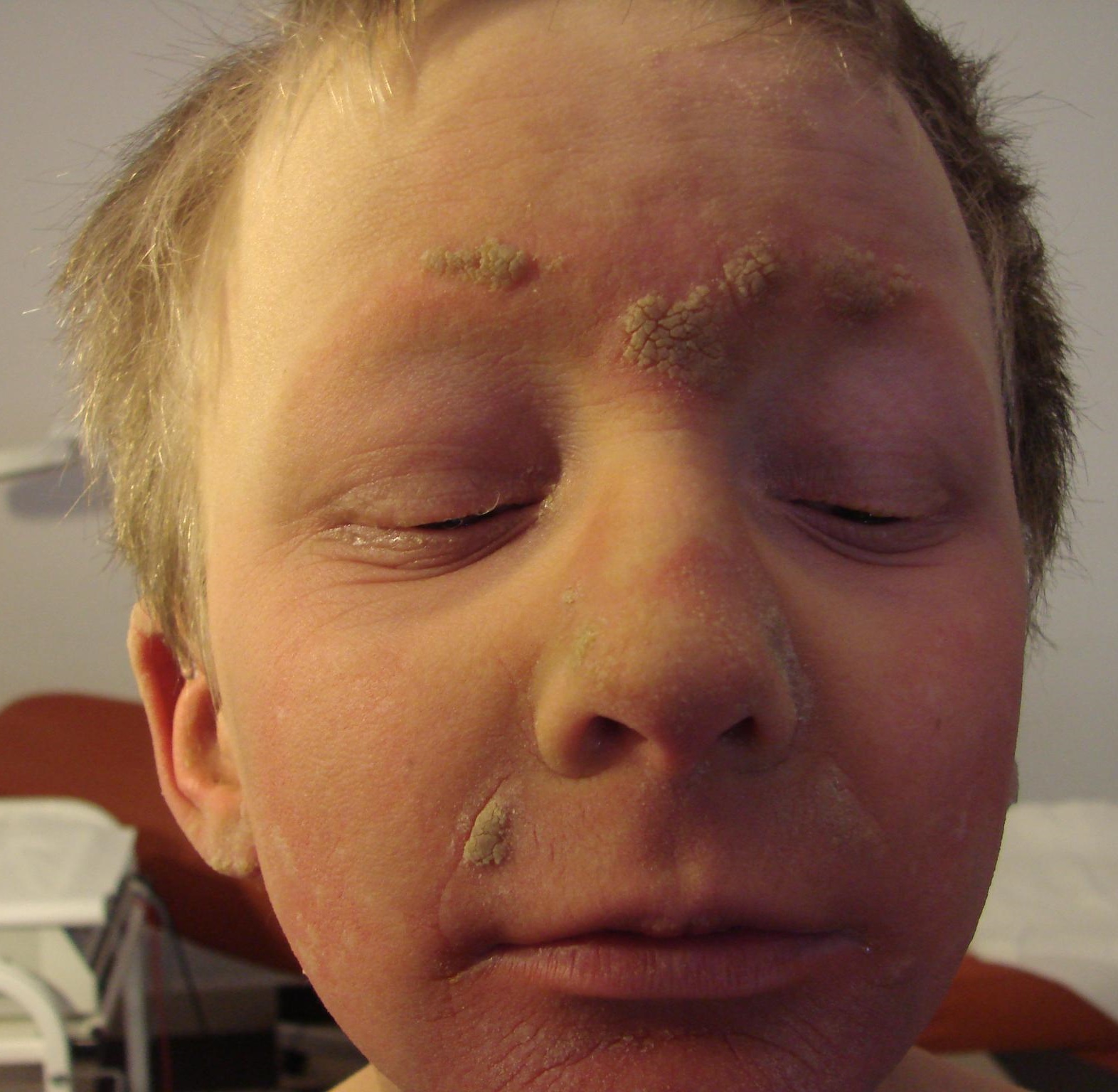
Subjectively patient did not report discomfort (except the cosmetic one) related with these skin changes nor did not complain for serious itching. Since early childhood patient had hearing problems that eventually led to partial deafness requiring the use of hearing aid apparatus. Genetic research revealed the evident presence of p.D50N gene and doubtful (within the margin of laboratory error) presence of a mother inherited GJB2 gene. Treatment included numerous otolaryngological consultations due to the slow but progressive hearing loss. Patient was treated with numerous topical preparations containing urea or salicylic acid to soften the ichtyotic skin. These preparations had an effect that was negligible or none. On the visit, the patient was prescribed a prescription for a preparation containing salicylic and benzoic acid, both in 10% concentration on fatty base (so called “Whitfield ointment”) to be administered to the most severe hyperkeratotic lesions.
Due to its congenital character, treatment of KID syndrome is very difficult and only supportive. Unfortunately, due to it’s severity, skin changes are very resistant to the topical treatment with keratolytic preparations. The patient described in the article had his hearing and vision partially affected. But in most severe cases chochlear implants and corneal transplants may be necessary. Treatment with oral antifungal agents is helpful in mucosal involvement with secondary fungal infection. Treatment with systemic retinoids that can improve affected skin status at least partially should be considered with care as they could worsen the status of already affected vision [7].
REFERENCES
1. Maintz L, Betz RC, Allam JP, Wenzel J, Jaksche A, Friedrichs N, et al. Keratitis-ichthyosis-deafness syndrome in association with follicular occlusion triad. Eur J Dermatol. 2005;15:347–52.
2. Gudmundsson S, Wilbe M, Ekvall S, Ameur A, Cahill N, et al. Revertant mosaicism repairs skin lesions in a patient with keratitis-ichthyosis-deafness syndrome by second-site mutations in connexin 26. Hum Mol Genet. 2017;26:1070-7.
3. Markova TG, Brazhkina NB, Bliznech EA, Bakhshinyan VV, Polyakov AV, Tavartkiladze GA. Phenotype in a patient with p.D50N mutation in GJB2 gene resemble both KID and Clouston syndromes. Int J Pediatr Otorhinolaryngol. 2016;81:10-4.
4. Coggshall K, Farsani T, Ruben B, McCalmont TH, Berger TG, Fox LP, et al. Keratitis, ichthyosis, and deafness syndrome: a review of infectious and neoplastic complications. J Am Acad Dermatol. 2013;69:127–34.
5. Bergman R, Mercer A, Indelman M, Sprecher E, Haim N, Zoller L, et al. KID syndrome: Histopathological, immunohistochemical and molecular analysis of precancerous and cancerous skin lesions. Br J Dermatol. 2012;166:455–7.
6. Jabłońska S, Chorzelski T. Choroby skóry –Warszawa, PZWL 1997.
7. Patel V, Sun G, Dickman M, Khuu P, Teng JM. Treatment of keratitis-ichthyosis- deafness (KID) syndrome in children: a case report and review of the literature. Dermatol Ther. 2015;28:89-93.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.