Association of palmoplantar and ocular keratoderma: Think of the Richner-Hanhart syndrome
Siham Boularbah , Zakia Douhi, Sabrina Oujdi, Mariam Soughi, Sara Elloudi, Hanane Baybay, Fatima-Zahra Mernissi
, Zakia Douhi, Sabrina Oujdi, Mariam Soughi, Sara Elloudi, Hanane Baybay, Fatima-Zahra Mernissi
Department of Dermatology, CHU Hassan II, Fez, Morocco
Citation tools:


Copyright information
© Our Dermatology Online 2023. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
A 28-year-old man was referred from ophthalmologists for management of his keratoderma. He was from first-degree marriage. He reported a painful palmoplantar (KPP) evolving since the age of 6 months. He reported also in the first few months of life resulting in photophobia, tearing, and redness. There were no similar cases in the family. Clinical examination showed striate keratoderma with linear hyperkeratosis on the dorsal overlaying extensor tendons with sharp demarcation (Fig. 1a). We also noted diffuse yellowish plantar and palmar hyperkeratosis sparing the instep with painful erosions (Figs. 1b and 1c). Ophthalmological examination revealed corneal opacification and strabism (Fig. 1d). Neurological examinations were without abnormalities. Considering the age of onset, corneal involvement, and KPP, Richner-Hanhart syndrome has been mentioned. Amino acid chromatography revealed an isolated increase in tyrosine. Richner-Hanhart syndrome was therefore retained. The patient has been placed on a restrictive diet with a daily proportion of 2 g/kg of protein with 400 mg/d of phenylalanine and tyrosine.
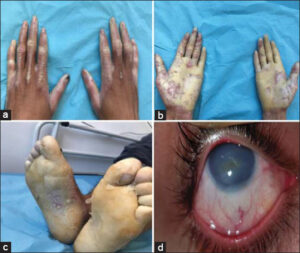 |
Figure 1: (a) Clinical image of dorsal hands showing striate keratoderma with linear hyperkeratosis on the dorsal overlaying extensor tendons with sharp demarcation. (b) Clinical image of palms showing yellowish keratoderma with painful erosions. (c) Clinical image of plants showing yellowish diffuse keratoderma with erosions sparing the instep. (d) Ophthalmological examination showing corneal opacification. |
Palmoplantar keratodermas (PPK) can be inherited or acquired. The major patterns of involvement are diffuse, striate, and punctata. They may be different on the hands and feet [1]. Patients suffer frequently from hyperhidrosis, maceration, pain, and fungal infections [1]. It is very important to determine the associated features such as nail dystrophy, hypotrichosis, dental anomalies, deafness, ocular findings, and cardiomyopathy. The association between PPK and ocular findings has to lead to Hanhart Richner’s disease [2]. This is a rare autosomal recessive disorder caused by a deficiency of tyrosine aminotransferase [3]. Clinical presentation of PPK is often focal in two major patterns: nummular type with oval and keratotic plaques mainly over pressure points; the striate type with linear hyperkeratotic lesions extending mostly from the palms to the volar surface of fingers. Focal PKK may evolve to the diffuse pattern over time like our observation [4]. Concerning ocular findings, there is a bilateral pseudodendritic keratitis simulating a herpetic origin which may lead to neovascularization and corneal scarring. The bilateral presentation of these ulcerations and the resistance to antivirals should alert the ophthalmologists and consider this diagnosis [5]. It can be often associated with cognitive impairment [6]. Plasma tyrosine concentrations are extremely high, and the eye and skin lesions probably result from the intracellular precipitation of tyrosine crystals. Dietary restriction of tyrosine and phenylalanine leads to the resolution of these lesions [2].
The diagnosis of Hanhart Richner disease should be established in the earlier year of life and the beginning of a restrictive diet low in phenylalanine and tyrosine [6]. These measures lead to a regression of PKK and ocular manifestations after a few months to avoid serious ocular or mental sequels.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Sakiyama T, Kubo A. Hereditary palmoplantar keratoderma “Clinical and genetic differential diagnosis“J. Dermatol.2016;43:264-74.
2. Zribi H. Richner-Hanhart Syndrome. Presse Med. 2016;45:264-5.
3. Dassouli R, Elloudi S, Choukri S, Baybay H, Douhi Z, Mernissi FZ. Dermoscopy of palmoplantar keratoderma:development and analysis methods. Our Dermatol Online. 2022;13:e44.
4. Bouyacoub Y, Zribi H, Azzouz H, Nasrallah F, Ben Abdelaziz R, Kacem M. Novel and recurrent mutations in the TAT gene in Tunisian families affected with Richner-Hanhart syndrome. Gene. 2013;529:45-9.
5. Kocabeyoglu S, Mocan MC, and Irkec M:In vivo confocal microscopic features of corneal pseudodendritic lesions in tyrosinemia type II. Cornea 2014;33:pp. 1106-1108.
6. Jen M, Nallasamy S. Ocular manifestations of genetic skin disorders. Clin Dermatol. 2016;34:242-75.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-3975-8316http://orcid.org/0000-0002-5942-441Xhttp://orcid.org/0000-0003-3455-3810 http://orcid.org/0000-0002-3975-8316http://orcid.org/0000-0002-5942-441Xhttp://orcid.org/0000-0003-3455-3810 |

Comments are closed.