Bardet–Biedl syndrome: Case report from a tertiary-care hospital in Srinagar, India
Saika Reyaz, Fozia Rehman , Shagufta Rather, Sheikh Javeed Sultan
, Shagufta Rather, Sheikh Javeed Sultan
Department of Dermatology, Venereology & Leprosy, Government Medical College Srinagar, Karan Nagar, Jammu and Kashmir, India
Citation tools:


Copyright information
© Our Dermatology Online 2023. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Bardet–Biedl syndrome (BBS) is a rare autosomal recessive ciliopathic disorder affecting multiple organ systems. The main clinical features are marked central obesity, retinal dystrophy, polydactyly, mental retardation, hypogonadism, and renal dysfunction. It affects both males and females. Herein, we report an interesting case of BBS with features of the BBS yet without cone-rod dystrophy, which is considered one of the hallmark features of this condition. To the best of our knowledge, this is the first report of Bardet–Biedl syndrome with the absence of cone-rod dystrophy and a normal fundus examination on the Indian subcontinent.
Key words: Bardet–Biedl syndrome; Ciliopathic disorder; Polydactyly
INTRODUCTION
Bardet–Biedl syndrome (BBS) is a rare autosomal recessive ciliopathic disorder, first described by Bardet and Biedl in 1920, characterized principally by marked central obesity, retinal dystrophy, polydactyly, mental retardation, hypogonadism, and renal dysfunction [1]. Its frequency varies from country to country, the incidence being much higher in some populations with a high level of consanguinity or those geographically isolated, with a disease incidence of 1 in 13,000 in the isolated populations of Newfoundland and Kuwait, 1 in 17,000 live births [2]. Mutations in at least 21 BBS genes have been recognized as causative factors [3]. Herein, we report an interesting case of BBS presenting to the dermatology outpatient with hypogenitalism and features such as marked central obesity, acanthosis nigricans, polydactyly, and mental retardation, yet with the absence of cone-rod dystrophy, hence the relevance of reporting this case with the absence of the hallmark feature.
CASE REPORT
A ten-year-old boy was reported to the skin outpatient department with complaints of asymptomatic darkening and thickening of the neck and both axillae, excessive weight gain, and underdevelopment of the genital organs. His mother revealed that he began to gain excessive weight since the age of five years. His background problems included diminished vision in both eyes. Regarding the birth history, he was second in birth order, born out of a second-degree, consanguineous marriage through normal vaginal delivery at full-term without complications. According to the mother, he had delayed motor and developmental milestones, with the commencement of walking and speech at the age of three and five, respectively. He was enrolled in a school by his parents yet, due to difficulty in learning, withdrew. There was a history of the death of one of the older male siblings soon after birth due to an unknown reason.
A physical examination revealed a rounded face with retrognathia, a high-arched palate, bilateral gynecomastia, and a protuberant abdomen (Fig. 1).
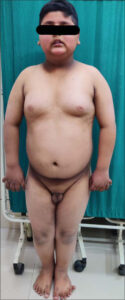 |
Figure 1: Ten-year-old boy with a rounded face, retrognathia, gynecomastia, central obesity, and underdeveloped genitalia. |
The height and weight were 135 cm and 65 kg, respectively, with a resultant body mass index of 36, which indicated severe obesity according to the revised consensus guidelines for India [4]. Blood pressure and the pulse rate were 110/70 millimeters of mercury and 88 per minute, respectively. Acanthosis nigricans was present in the axillary and neck regions (Fig. 2a).
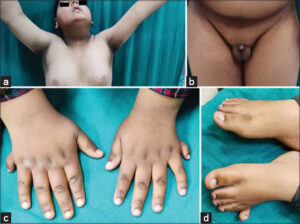 |
Figure 2: (a) Acanthosis nigricans in both axilla and neck regions. (b) Hypogonadism with a micropenis. (c) Polydactyly with hexadactyly of the hands (d) Hexadactyly of the right foot. |
Dermoscopy of acanthosis nigricans in the neck region was performed, which demonstrated the presence of crista cutis and sulci cutis on a diffuse, dark brown background (Fig. 3).
 |
Figure 3: Dermoscopic image of acanthosis nigricans on the neck (DermLite; DL4; 10×) showing brown dots (blue arrow), linear crista cutis, and sulci cutis (yellow and black arrow, respectively) against a dark brown background. |
Microtestis with a testicular volume of 1.5 mL (normal: 10–12 mL) as documented by ultrasonography and microphallus (< 2.5 cm) were observed (Fig. 2b).
Extra-axial-polydactyly was present in both upper limbs and the right foot with a normal digit number on the left foot (Figs. 2c and 2d).
A psychiatric consultation revealed that his mental development lagged behind the normal range, with an IQ below seventy.
On the ophthalmological examination, visual acuity was 18/36 in both eyes. However, a fundus examination was normal.
A neurological examination and auditory assessment were found to be normal.
Laboratory investigations, including a hemogram, liver function tests, renal function tests, urine analysis, chest X-ray, electrocardiogram, and echocardiogram were normal. Ultrasound revealed grade-2 fatty liver disease. A fasting lipid profile revealed an increased level of triglycerides (TG)—200 mg/dL (normal: 32–100 mg/dL)—and a decreased level of high density lipoprotein (HDL)—24 mg/dL (normal for males: 40 mg/dL). Serum cortisol levels were normal. Genetic analysis was not conducted as it was not available in our hospital.
We diagnosed the case as BBS as our patient met four primary clinical features along with two secondary features based on the diagnostic criteria described below (Table 1).
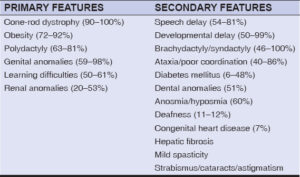 |
Table 1: Diagnostic criteria of BBS [1]. |
As there is no definitive treatment for BBS, we counseled the child’s parents about the genetic basis of the disease. We advised our patient to increase physical activity in the form of regular walks for thirty minutes a day at least four times per week, and a weight-reducing diet was advised as per the dietician’s recommendations. Spectacles were advised by ophthalmology for low vision. The patient was advised to return for regular follow-up to observe any progressive ophthalmological changes and for behavioral therapy.
DISCUSSION
Bardet–Biedl syndrome is a rare autosomal recessive ciliopathic disorder named after George Louis Bardet, a French physician, and Arthur Biedl, a Hungarian pathologist and endocrinologist. There was a debate in the medical literature regarding the condition reported by Lawrence and Moon in 1886, referred to as Laurence–Moon syndrome (LMS). Laurence–Moon–Bardet–Biedl syndrome (LMBBS) is an abandoned term as patients with Laurence–Moon presented with spastic paraplegia with no polydactyly and obesity, which are the primary features of BBS. Hence, BBS and LMS are regarded as separate entities.
The underlying pathology of BBS remains unclear. The basic reason for this pleiotropic disease originates from cilia dysfunction. Hence, the condition falls under the spectrum of “ciliopathies” [5]. Mutations in 21 BBS genes (BBS1–BBS20 and NPHP1) have been cloned, all of these genes having a relationship with cilia biogenesis or function [3].
According to these criteria, four primary or three primary and two secondary criteria are sufficient for the diagnosis. Four primary and two secondary features were present in our patient (shown in bold), thus fulfilling the diagnostic criteria of BBS.
The presence of cone-rod dystrophy is one of the hallmark clinical features of BBS. Decreased vision has been reported in the first decades of life in patients with BBS, as was in our case, with the majority legally blind (best corrected visual acuity < 6/60) by the second or the third decade of life [6].
Obesity is another cardinal feature in BBS, with a prevalence of 72–92%. The cause of obesity is 1) the deregulation of appetite; 2) impaired leptin receptor signaling; 3) a reduced number of cilia due to BBS gene mutations [7]. Obesity is usually noticed by parents within the first ten years of life in comparison to other children and playmates of similar age.
Limb anomalies such as polydactyly and syndactyly are seen since birth, thus providing a useful diagnostic clue for BBS as 63–81% of patients manifest the same [5].
Hypogonadism manifests as hypogenitalism in males and genital anomalies in females, with a delayed onset of puberty in both sexes. A small penis buried in the adipose tissue and a decreased testis volume are often seen in male patients.
Around 44% of cases with BBS have a learning disability, with an IQ level of 79 or below.
Renal anomalies seen in BBS include structural anomalies, hydronephrosis, vesicoureteral reflux, and progressive renal parenchymal disease, which is commonly associated with urinary concentration defects [8]. Chronic kidney disease (CKD) is a major contributor of morbidity and mortality in individuals with BBS.
An effective multidisciplinary approach is required to manage this pleiotropic condition. There should be an awareness of complications, for which BBS has laid the base and patients should be followed up in this respect. Both a proper diet (low in calories, low in protein), exercise programs, periodic vision evaluation, and up-to-date changing of their prescription lenses should be encouraged.
CONCLUSION
Bardet–Biedl syndrome is an autosomal recessive inherited disorder with wide variability in expression and a disease of genetic complexity. To the best of our knowledge, this is the first report of Bardet–Biedl syndrome with the absence of cone-rod dystrophy and a normal fundus examination on the Indian subcontinent. A timely and thorough management plan by a multidisciplinary team should allow these children to integrate better into society and thrive fully. Furthermore, both parents should undergo genetic counseling, especially those with a history of consanguineous marriages in the family.
CONSENT
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Beales PL,Elcioglu N, Woolf AS, Parker D, Flinter FA. New criteria for improved diagnosis of Bardet–Biedl syndrome:Results of a population survey. J Med Genet. 1999;36:437-46.
2. Moore SJ, Green JS, Fan Y, Bhogal AK, Dicks E, Fernandez BA, et al. Clinical and genetic epidemiology of Bardet–Biedl syndrome in Newfoundland:A 22-year prospective, population-based, cohort study. Am J Med Genet.2005;132:352-6.
3. Suspitsin EN, Imyanitov EN. Bardet–Beidl syndrome. Molecular Syndromol.2016;7:6271.
4. Misra A, Chowbey P, Makkar BM, Vikram NK, Wasir JS, Chadha D, et al;Concensus Group. Consensus statement for diagnosis of obesity, abdominal obesity and the metabolic syndrome for Asian Indians and recommendations for physical activity, medical and surgical management. J Assoc Physicians India. 2009;57:163-70.
5. E. Forsythe E, Beales P.L, Bardet–Biedl syndrome. Eur J Hum Genet.2013;21:8-13.
6. Mockel A, Perdomo Y, Stutzmann F, Letsch J, Marion V, Dollfus H. Retinal dystrophy in Bardet–Biedl syndrome and related syndromicciliopathies. Prog Retin Eye Res. 2011;30:258-74.
7. Guo DF, Rahmouni K. Molecular basis of the obesity associated with Bardet–Biedl syndrome. Trends Endocrinol Metab. 2011;22:286-93.
8. Putoux A, Attie-Bitach T, Martinovic J, Gubler M-C. Phenotypic variability of Bardet–Biedl syndrome:Focusing on the kidney. Paediatr Nephrol.2012;27:7-15.
Notes
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-0980-6014 http://orcid.org/0000-0002-0980-6014 |

Comments are closed.