Dowling-Degos disease: An association with hidradenitis suppurativa
Abdullah Mancy
Department of Dermatology and Venereology, Al-Ramadi Teaching Hospital, Al-Anbar Health Directorate, Ministry of Health/Iraq
Citation tools:

Copyright information
© Our Dermatology Online 2022. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Dowling-Degos disease is one of the genodermatoses presenting with acquired reticulate pigmentation of the flexures, black-dot papules, and pitted scars. Numerous associated conditions and multiple variants of the disease have been reported in the literature. Several gene mutations play a role in the pathogenesis of the disease giving rise to multiple phenotypic expressions. Herein, we discuss a case in three generations of a family affected with the disease and shed light on the associations and various expressions of the disease.
Keywords: Reticulate pigmentation; Hidradenitis suppurativa; Autosomal dominant inheritance
INTRODUCTION
Dowling-Degos disease (DDD), also termed reticular pigmented anomaly of the flexures, is a rare genetic disease with an autosomal dominant mode of inheritance with different phenotypic expressions [1]. It usually presents after puberty with reticular pigmentation of the flexures, particularly the axillae, groin, and submammary folds, blackhead comedo-like papules, and perioral, acneiform, pit-like scars [2]. DDD results from a mutation in more than one gene [3]. It is associated with numerous conditions, such as hidradenitis suppurativa (HS) [4], multiple epidermal cysts [5], keratoacanthoma [6], squamous cell carcinoma, and malignant melanoma [7].
CASE REPORT
A 39-years old diabetic male presented with a painful ulcer on the posterior surface of the upper part of the right thigh persistent for five days. An examination revealed an irregular ulcer, around 1.5 × 2 cm in diameter, with its edge macerated, around 2 cm in depth, and with a reddish base. It was surrounded by an erythematous zone, mostly representing a ruptured cyst (Fig. 1a). There were also multiple cysts, nodules, and scars resembling HS on the buttocks persistent for fifteen years (Fig. 1b). Similar lesions were located on the back of the scalp and neck (Fig. 1c). In addition to these lesions, multiple pitted, acneiform scars were present on the face, involving the nose, paranasal areas, nasal bridge, cheeks, and forehead (Fig. 2a). Reticulated confluent hyperpigmented patches and discrete macules at its periphery, hyperpigmented keratotic papules, black, comedo-like lesions, and multiple linear, atrophic scars were present in the axillae and groin, with the presence of a solitary cyst in the right axilla (Figs. 2b – 2e). Also, numerous atrophic scars and blackhead comedones were found on the upper back and dorsal surfaces of both elbows (Figs. 3a – 3c). These skin features appeared at the age of fifteen years. The hair, nail, and mucus membranes were normal. The family history was positive for other members of the family (Fig. 4). A histopathological examination of the pigmented macule on the axilla revealed a normal stratum corneum with a basket-weave pattern. The other layers of the epidermis were slightly thin. There was a suprapapillary thinning of the epidermis. The rete ridges were thin, branched, elongated, and with basal pigmentation, with an antler-like appearance. There was mild lymphocytic infiltration of the dermis and some melanophages (Fig. 5). The patient was treated with systemic and topical antibiotics as an acute treatment for the ruptured, infected cyst. Thus, a diagnosis of DDD was reached based on the clinical manifestations, mode of inheritance, and histopathological characteristics. Informed consent was taken from the patient and the study was approved by the ethical committee.
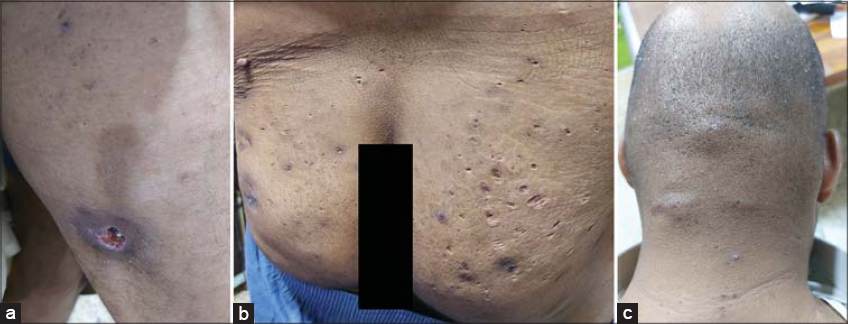 |
Figure 1: (a) Ulcer on the upper part of the thigh. (b) Multiple atrophic scars and cysts on the buttock. (c) Multiple cysts on the back of the scalp and scars on the back of the neck. |
| Figure 2: (a) Multiple pitted, acne-like scars on the face. (b) Reticulated hyperpigmentation of the right axilla with a cyst. (c) Reticulated hyperpigmentation of the left axilla. (d-e) Reticulated hyperpigmentation of the right and left groin. | |
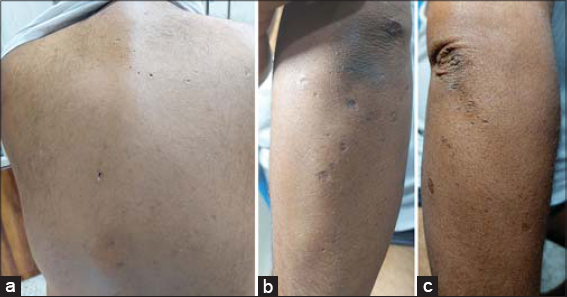 |
Figure 3: (a) Multiple pitted, acne-like scars on the back. (b-c) Multiple atrophic scars of the elbows. |
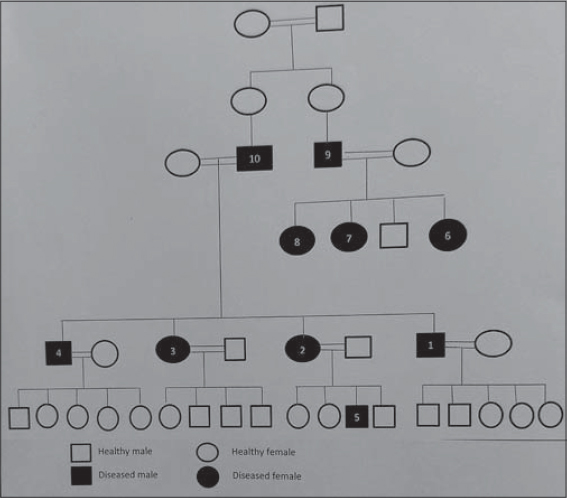 |
Figure 4: Family pedigree. |
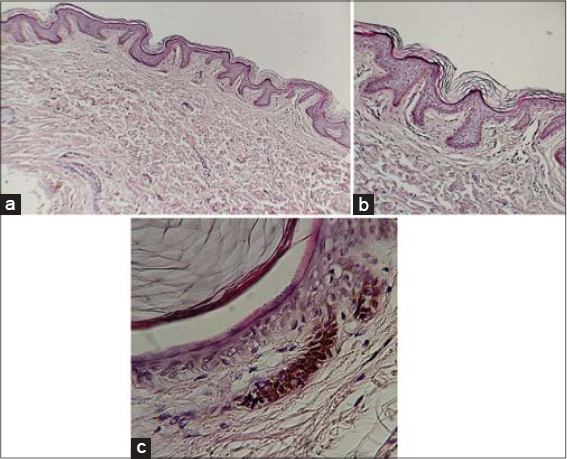 |
Figure 5: Histopathological examination of the hyperpigmented macule on the axilla (a: H&E, 4×; b: 10×; c: 40×), revealing branched, elongated, and antler-like rete ridges, basal hyperpigmentation, and mild perivascular lymphocytic infiltrate. |
DISCUSSION
DDD (OMIM #179850) is a genetic dermatosis manifesting with discrete, light brown or black, acquired macules arranged in a reticular form [7,8]. It usually appears first in the intertriginous areas, especially the axillae and groin, then progresses slowly [9]. In most reported cases, the involvement is of the face with acneiform pitted scars, usually located around the mouth [2,10,11]. In this case, most of the face was involved, the nose, paranasal areas, nasal bridge, forehead, and cheeks, while the perioral areas were spared. These manifestations were also observed by Gupta et al. [7] and Singh et al. [12]. This could be explained by a certain genetic mutation giving rise to this specific phenotypic expression.
DDD usually appears after puberty and, although inherited as autosomal dominant, females are more frequently affected [10,13]. In this family, the disease appeared at an age of fifteen years, thus an earlier onset may be determined by a specific mutation, although the disease was reported even in a newborn by Zuo et al. [10].
DDD results from mutations of more than one gene [3]. Such mutations affect keratin 5 (KRT5) and the POFUT1, POGLUT1, and PSENEN genes [4,9,14]. The PSENEN gene is linked to DDD associated with HS [14]. The POFUT1, POGLUT1, and PSENEN genes ameliorate the NOTCH signaling pathway, which is essential in keeping the integrity of the hair follicle sheath and other skin appendages [3,10,15,16]. It also plays an essential role in the maintenance of the homeostasis of cutaneous cells and controls the growth, maturation, and differentiation of keratinocytes and melanocytes [14].
Numerous dermatoses are associated with DDD [5–7]. One of these is HS, in which the involvement of the follicular unite is an important step in its pathogenesis [4,5]. It has been found that defects in the NOCH signaling pathway lead to disturbances in epidermal and follicular maturation [17]. The POFUT1 and POGLUT1 genes play a role in the regulation of the NOTCH pathway [18]. Hence, interactions among these different pathways may result in the appearance of HS in this disease. In our case, there was the involvement of the back of the neck and scalp with multiple cysts and previous scars. As recently described by Eugenia et al., the involvement of the nape area in a patient with HS is more likely to be associated with DDD [19]. Some diseases ought to be differentiated from DDD, such as Haber’s syndrome, acanthosis nigricans, neurofibromatosis type 1, prurigo pigmentosa, Galli-Galli disease, and the follicular variant of DDD, reticulate acropigmentation of Kitamura, acropigmentation of Dohi, and dyschromatosis universalis hereditarian [8,11,13], yet clinical and histopathological criteria may exclude these dermatoses.
Numerous options are employed for the treatment of the disease, such as the depigmenting agents hydroquinone and retinoids. Also, laser is employed, such as Er: YAG, and a combination of Q-switched Nd: YAG and fractional CO2 lasers, yet these modalities produce limited effects [20].
CONCLUSION
As reported in the literature, DDD resembles a spectrum of diseases, not one single, with localized and generalized forms at the ends of the spectrum, in between numerous varieties and associations. An early-age onset, the involvement of the nape of the neck and the back of the scalp, sparing of the perioral areas in some members, and association with HS in others, require genetic studies to detect the underlying defects giving rise to the variable phenotypic expressions.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Taeeb A, Morice-Picard F, Ezzedine K. Genetic disorders of pigmentation. In:Barker J, Bleiker T, Chalmers R, Creamer D, Griffiths C, editors. Rook’s Textbook of Dermatology. 9th ed. Oxford:Wiley Blackwell;2016 p. 1855-1873.
2. Naveen KN, Athaniker SB, Hegde SP, Shetty R, Radha H, Parinitha SS. Atypical cases of DowlingDegos disease. Indian Dermatol Online J. 2016;7:99-102.
3. George A, George R, Mathew AJ, Telugu RB. DowlingDegos disease with hidradenitis suppurativa and inflammatory arthritis in two generations. Indian Dermatol Online J. 2020;11:413-5.
4. Arjona-Aguilera C, Linares-Barrios M, Albarrán-Planelles C, Jiménez-Gallo D. DowlingDegos disease associated with hidradenitis suppurativa:A case report. Actas Dermosifiliogr. 2015;106:337-8.
5. Loo J, Rytina E, Todd PM. Hidradenitis suppurativa, DowlingDegos and multiple epidermal cysts:A new follicular occlusion triad. Clin Exp Dermatol. 2004;29:622-4.
6. Fenske NA, Groover CE, Lober CW, Espinoza CG. DowlingDegos disease, hidradenitis suppurativa, and multiple keratoacanthomas:A disorder that may be caused by a single underlying defect in pilosebaceous epithelial proliferation. J Am Acad Dermatol. 1991;24:888-92.
7. Gupta V, Sahni K, Khute P, Sharma VK, Ali MF. DowlingDegos disease and malignant melanoma:Association or mere coincidence? Indian J Dermatol Venereol Leprol. 2015;81:627-8.
8. James WD, Rosenbach MA, Elstone DM, Treat JR, Neuhaus IM. Disturbances of pigmentation. In:Andrews Diseases of the Skin:Clinical Dermatology.13th ed. Elsevier, 2020;991-1012.
9. Knuever J, Persa OD, Illerhaus A, Ralser DJ, Hartmann K, Betz RC, et al. Mast cell activation in Dowling-Degos disease. Br J Dermatol. 2019;181:1312-14.
10. Zuo YG, Ho M, Jin HZ, Wang BX. Case report:A case of newborn onset reticulate pigmented anomaly of the flexures in a Chinese female. J Eur Acad Dermatol Venereol. 2008;22:901-3.
11. El Kadiri S, Bay Bay H, Chaoui R, Douhi Z, Elloudi S, Mernissi FZ. A rare classic case of Dowling-Degos disease with dermoscopy description. Our Dermatol Online. 2020;11:e71.1-e71.2.
12. Singh S, Khandpur S, Verma P, Singh M. Follicular Dowling Degos disease:A rare variant of an evolving dermatosis. Indian J Dermatol Venereol Leprol. 2013;79:802-04.
13. Horner ME, Parkinson KE, Kaye V, Lynch PJ. Dowling-Degos Disease involving the vulva and back:Case report and review of the literature. Dermatol Online J. 2011;17:1.
14. Ralser DJ, Basmanav FB, Tafazzoli A, Wititsuwannakul J, Delker S, Danda S et al. Mutations in g-secretase subunit-encoding PSENEN underlie Dowling-Degos disease associated with acne inversa. J Clin Invest. 2017;127:1485-90.
15. Li M, Cheng R, Yao Z. Mutations in POFUT1, encoding protein O-fucosyltransferase1, cause generalized Dowling-Degos disease. Am J Hum Genet. 2013;92:895-903.
16. Kiuru M, Terrell J, Gorouhi F, McPherson J. Novel POFUT1 mutation in patient with flexural and acral hyperpigmented reticulated macules presenting in adolescence. JAAD. 2020;6:334-6.
17. Wang B. Secretase mutation and consequently immune reaction involved in pathogenesis of acne inversa. J Investig Dermatol Symp Proc. 2015;17:25.
18. Kelly G, Sweeney CM, Tobin AM, Kirby B. Hidradenitis suppurativa: The role of immune dysregulation. Int J Dermatol. 2014;53:1186-96.
19. Eugenia AB, Iris GV, Jorge R, Jose CP, Miquel RP, Jesús E. DowlingDegos disease and hidradenitis suppurativa:Epidemiological and clinical study of 15 patients and review of the literature. Acta Derm Venereol. 2019;99:917-8.
20. Tambe SA, Patil PD, Saple DG. Successful management of DowlingDegos disease with combination of Q-switched Nd:YAG and fractional carbon dioxide laser. J Cutan Aesthet Surg. 2017;10:60-2.
Notes
Source of Support: Nil,
Conflict of Interest: The authors have no conflict of interest to declare.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-3688-6953 http://orcid.org/0000-0002-3688-6953 |

Comments are closed.