Endometrial cancer in neurofibromatosis type I (von Recklinghausen’s disease): A case report and literature review
Justin Bagorane 1, Gustave Negamiyimana1, Germaine Niyitanga2, Adam Hajjine2, Mohamed El Fadli1, Rhizlane Belbaraka1
1, Gustave Negamiyimana1, Germaine Niyitanga2, Adam Hajjine2, Mohamed El Fadli1, Rhizlane Belbaraka1
1Department of Medical Oncology, Mohamed VI Hospital, University; Cadi Ayyad University, Marrakech, Morocco, 2Department of Radiology, Mohamed VI Hospital University; Cadi Ayyad University, Marrakech, Morocco
Corresponding author: Justin Bagorane, MD
How to cite this article: Bagorane J, Negamiyimana G, Niyitanga G, Hajjine A, El Fadli M, Belbaraka R. Endometrial cancer in neurofibromatosis type I (von Recklinghausen’s disease): A case report and literature review. Our Dermatol Online. 2022;13(2):179-182.
Submission: 12.08.2021; Acceptance: 23.01.2022
DOI: 10.7241/ourd.20222.15
Citation tools:

Copyright information
© Our Dermatology Online 2022. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Neurofibromatosis type I is a complex, multisystem cancer predisposition syndrome of a number of tumors, which commonly arise in the central and peripheral nervous system, the gastrointestinal tract (GIT), and breast and soft tissues, yet the association with endometrial cancer is rare. Herein, we report the case of a 67-year-old female, never married, who had been followed since puberty for neurofibromatosis type I clinically manifested by pigmentary abnormalities such as café-au-lait macules, neurofibromas, and Lisch nodules with various comorbidities, such as diabetes type I, hypertension, renal failure, and ophthalmic disorders (poor vision). One year earlier, she developed a locally advanced endometrial cancer, for which chemotherapy would not have been optimal because of these comorbidities; due to the pathology of neurofibromatosis, the lifespan of such individuals is typically around fifteen years shorter than that of the general population and it negatively impacts their quality of life.
Key words: Neurofibromatosis type I; Cutaneous neurofibroma; Endometrial cancer
INTRODUCTION
Neurofibromatosis type I (NF-1), also known as von Recklinghausen’s disease, is a complex, multisystem cancer predisposition syndrome, with a birth incidence falling between 1 in 2500 and 1 in 3000 individuals worldwide [1]. The syndrome is caused by autosomal dominant inheritance or the loss of function by de novo mutations in the NF-1 tumor suppressor gene located on chromosome 17 (17q11.2) [2]. The diagnosis of NF-1 is based on clinical manifestations, including pigmentary abnormalities, peripheral and central nervous tumors, bone abnormalities, vasculopathy, and other cancers [3]. The lifespan of individuals with NF-1 is typically around fifteen years shorter than that of the general population [4]. In large part, this shortened lifespan reflects the fact that patients with NF-1 are at an increased risk of developing several types of benign and malignant neoplasms. Although a number of these tumors arise in the central nervous system and the peripheral nervous system (PNS), several types of NF-1-associated neoplasms occur elsewhere, including the skin, the gastrointestinal tract, bone marrow, the breasts, and soft tissues [5]. Gynecological cancers, such as endometrial adenocarcinoma, are not commonly encountered in NF-1.
CASE REPORT
Herein, we report the case of a 67-year-old female, never married, who had, since puberty, presented a neurofibromatosis (NF1) manifested by pigmentary abnormalities such as café-au-lait macules and various sizes of cutaneous neurofibromas (Figs. 1a and 1b). The patient had been treated for diabetes and hypertension for more than fifteen years. There was no family history of the same clinical case.
 |
Figure 1: (a and b) Clinical manifestations by pigmentary abnormalities such as café-au-lait macules and various sizes of cutaneous neurofibromas in an adult with von Recklinghausen’s disease. |
She had been followed at the oncology department since July 2020 for well-differentiated, infiltrating endometrioid adenocarcinoma diagnosed at the gynecology department because of heavy postmenopausal bleeding.
A thoracic and abdominopelvic CT (TAP-CT) scan performed on August 18, 2020, revealed a well-limited, hypodense tumor of the uterine cavity, measuring 88 × 108 × 123 mm (AP × T × CC) in size, enhanced heterogeneously after the injection of a contrast product, locally infiltrating the posterior face of the bladder, coming into contact with the lower rectum and the recto-sigmoid hinge with the loss of the fatty separation border in places behind, and arriving in contact with the slender handles with the loss of the fatty separation border above and internal bilateral and external right iliac lymphadenopathy. The tumor has been classified as cT4N2M1, stage IVA (Figs. 2 and 3).
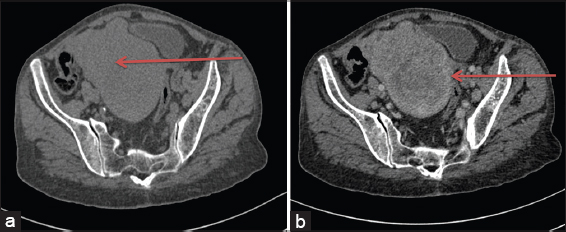 |
Figure 2: (a and b) Axial CT scan section showing a tumor of the uterine body, well-limited, spontaneously isodense, and enhancing heterogeneously after the injection of a contrast product. |
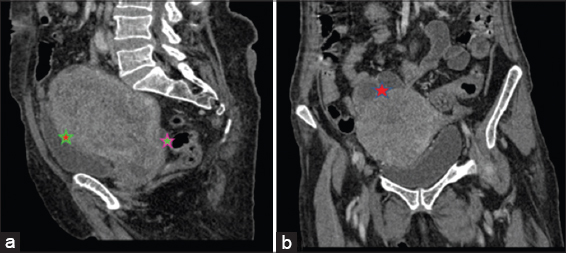 |
Figure 3: (a and b) Sagittal and coronal CT scan sections showing that the tumor came into contact with the bladder in the front and the lower rectum behind (a), and into contact with the slender handles above (b). |
She received chemotherapy treatment based on the combination of paclitaxel 175 mg/m2 and carboplatin AUC5 (day 1 = day 21). An evaluation after three cycles of chemotherapy found a progression in the size of the tumor. She benefited from a second line of chemotherapy based on doxorubicin 60 mg/m2 and carboplatin AUC4. She was unfit on cisplatin due to a renal insufficiency, with clearance on creatinine at 47.6 ml/min and received a partial radiological response during an evaluation after four cycles of chemotherapy.
DISCUSSION
The diagnosis of neurofibromatosis type I is commonly based on clinical assessment according to the diagnostic criteria formulated by the National Institutes of Health Consensus Development Conference [6], underlining skin manifestations and the bone and nervous system. (Table1). Occasionally, other signs of NF-1 may not develop until the late teens or early twenties, and slit-lamp examination against Lisch nodules may be helpful in these patients. NF-1 mutational analysis clarifies the diagnosis in some uncertain cases. However, genetic testing is not advocated routinely and expert consultation is advised before it is undertaken. Furthermore, a biopsy of asymptomatic cutaneous neurofibromas should not be undertaken for diagnostic purposes in individuals with clear-cut NF-1 [7]. Cutaneous neurofibromas (cNFs) are among the most common manifestations of NF-1, affecting around 99% of patients with NF-1 [7]. They are unlikely to undergo malignant transformation or to cause severe neurologic disabilities or fatal complications. Nevertheless, cNFs are considered one of the greatest concerns in patients with NF-1, especially adults. These concerns are mainly due to disfigurement and dysesthesia, causing substantial psychological distress and negative body image perception [8]. In our case, the patient was 67-year-old, never married, did not continue her studies as her generation, which negatively impacted her quality of life (Table 1).
 |
Table 1: Diagnostic criteria for neurofibromatosis type I (NF-1). |
The treatment of cNFs involves monitoring or procedure-based therapy. Although surgical resection allows the complete removal of the lesion, there are obstacles, including a limited number of lesions that may be treated in one session and possible scars. Other alternatives include electrodessication, which removes cNFs through dehydration and denaturation [9]. This allows for the removal of large numbers of cNFs in a single session, yet requires general anesthesia and may cause scarring and pigmentation changes. Other procedure-based therapies for cNFs are laser photocoagulation [10] and radiofrequency ablation [11]. Another approach uses a local drug in photodynamic therapy, which is being tested for different cancers [12]. The use of ketotifen is one of the first managed efforts for the treatment of cNFs and their associated symptoms [13]. Ketotifen is used to block the histamine 1 receptor, which helps to stabilize mast cells; its use in NF-1 is based mostly on the finding of abundant mast cells in neurofibromas. A significant improvement in pain and pruritus has been reported, but an objective tumor reduction has not been documented [14]. As for the local therapeutic approaches, some drugs have been tested for cNFs with promising results, such as ranibizumab, which is a vascular endothelial growth factor monoclonal antibody injected intralesionally, imiquimod, which shows minimal efficacy in tumor shrinkage compared to the baseline volume, and topical rapamycin, an mTOR inhibitor [14]. The safety profile of these tested drugs remains a major concern for physicians, regulators, patients, and their caregivers. Because of our patient’s poor financial situation, she had no chance to benefit from these emergent treatments for her cutaneous neurofibromas, which had impacted the quality of her life.
The NF-1 gene encodes neurofibromin, which has been shown to control cell growth through two major intracellular pathways. First, neurofibromin regulates negatively RAS pathway signaling through its action on GTPase-activating protein (GAP), stimulating the conversion of GTP-bound RAS to its GDP-bound form [15]. Increased RAS activity leads to the downstream activity of the MEK/ERK pathway as well as the PI3K/AKT/mTOR pathway [16]. This genetic deregulation in females with neurofibromatosis type I may explain the exceptionally high risk of developing endometrial cancer, in which the PI3K/AKT/mTOR pathway is the most frequently deregulated pathway via mutations in PTEN and/or PIK3CA [17].
NF-1 associated with an endometrial cancer is poorly documented in the literature. Optimal oncological management by chemotherapy is often compromised following the coexistence of various comorbidities in NF-1, as in our case, which was diabetic and hypertensive, had a renal insufficiency, and had repetitive episodes of neutropenia and anemia that required multiple blood transfusions.
CONCLUSION
In conclusion, neurofibromatosis type I is inherited in an autosomal dominant manner. Around half of individuals with NF-1 have been found to have an affected parent, and more than 50% have an altered gene as a result of de novo mutation. With NF-1 is suspected in a child, its parents should have medical histories, physical examinations, and ophthalmological slit-lamp examinations performed with particular attention to the features of NF-1. In females older than 60 years, the annual performance of endovaginal ultrasound would allow the diagnosis and early management of an associated endometrial cancer. A diagnosis of NF-1 in a family member may permit an unequivocal diagnosis of NF-1 in a child; it is essential and highly important for genetic counseling and in medical implications for the affected parent.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Evans DG, Howard E, Giblin C, Clancy T, Spencer H, Huson SM, et al. Birth incidence and prevalence of tumor-prone syndromes:Estimates from a UK family genetic register service. Am J Med Genet Part A. 2010;152:327-32.
2. KallionpääRA, Uusitalo E, Leppävirta J, Pöyhönen M, Peltonen S, Peltonen J. Prevalence of neurofibromatosis type 1 in the Finnish population. Genet Med. 2018;20:1082-6.
3. Jett K, Friedman JM. Clinical and genetic aspects of neurofibromatosis 1. Genet Med. 2010;12:1-11.
4. Rasmussen SA, Yang Q, Friedman JM. Mortality in neurofibromatosis 1:An analysis using U.S. death certificates. Am J Hum Genet. 2001;68:1110-8.
5. Longo JF, Weber SM, Turner-Ivey BP, Carroll SL. Recent advances in the diagnosis and pathogenesis of neurofibromatosis type 1 (NF1)-associated peripheral nervous system neoplasms. Adv Anat Pathol. 2018;25:353-68.
6. National Institutes of Health Consensus Development Conference statement:Neurofibromatosis. Arch Neurol. 1988;45:575-8.
7. Ferner RE, Gutmann DH. Neurofibromatosis type 1 (NF1):Diagnosis and management. 1re éd. Vol. 115, Handbook of Clinical Neurology. Elsevier B.V.;2013. 939-55.
8. Patricia Z. Page GPP, Emmanuel Ecosse BRK, Allain Leplege and PW. Impact of neurofibromatosis 1 on quality of life. Am J Med Genet. 2006;221:212-21.
9. Lutterodt CG, Mohan A, Kirkpatrick N. The use of electrodessication in the treatment of cutaneous neurofibromatosis:A retrospective patient satisfaction outcome assessment. J Plast Reconstr Aesthetic Surg. 2016;69:765-9.
10. Elwakil TF, Samy NA, Elbasiouny MS. Non-excision treatment of multiple cutaneous neurofibromas by laser photocoagulation. Lasers Med Sci. 2008;23:301-6.
11. Kim SH, Roh SG, Lee NH, Yang KM. Radiofrequency ablation and excision of multiple cutaneous lesions in neurofibromatosis type 1. Arch Plast Surg. 2013;40:57-61.
12. Dolmans DEJG., K.Jain DF and R. Photodynamic therapy for cancer. Nat Rev cancer. 2003;3:375-80.
13. Riccardi VM. Ketotifen suppression of NF1 neurofibroma growth over 30 years. Am J Med Genet Part A. 2015;167:1570-7.
14. Lobbous MR. Korf B. Therapeutic development in neurofibromatosis. Neurofibromatosis – Curr Trends Futur Dir. 2020. IntechOpen.
15. Basu TN, Gutmann DH, Fletcher JA, Glover TW, Collins FS, Downward J. Aberrant regulation of ras proteins in malignant tumour cells from type 1 neurofibromatosis patients. Nature. 1992;359:167-9.
16. Ratner N, Miller SJ. A RASopathy gene commonly mutated in cancer:The neurofibromatosis type 1 tumour suppressor. Nat Rev Cancer. 2015;15:290-301.
17. Catasus L, Gallardo A, Cuatrecasas M, Prat J. Concomitant PI3K-AKT and p53 alterations in endometrial carcinomas are associated with poor prognosis. Mod Pathol. 2009;22:522-9.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0003-0563-841X http://orcid.org/0000-0003-0563-841X |

Comments are closed.