Bullous lupus erythematosus with basement membrane deposits of IgD
Ana Maria Abreu Velez 1, Bruce R Smoller2,3, Michael S. Howard1
1, Bruce R Smoller2,3, Michael S. Howard1
1Georgia Dermatopathology Associates, Atlanta, Georgia, USA, 2,3Department of Pathology and Laboratory Medicine, University of Rochester School of Medicine and Dentistry, Rochester, New York, USA, 3Department of Dermatology, University of Rochester School of Medicine and Dentistry, Rochester, New York, USA
Corresponding author: Ana Maria Abreu Velez, M.D., Ph.D.
How to cite this article: Abreu Velez A, Smoller BR, Howard MS. Bullous lupus erythematosus with basement membrane deposits of IgD. Our Dermatol Online. 2022;13(2):161-164.
Submission: 07.12.2021; Acceptance: 05.02.2022
DOI: 10.7241/ourd.20222.10
Citation tools:

Copyright information
© Our Dermatology Online 2022. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Bullous lupus erythematosus is an uncommon blistering subepidermal autoimmune disease with characteristic immunopathological nosologic features. A 67-year-old female presented with the sudden appearance of blisters in the upper area of the right chest. Skin biopsies stained with H&E favored the diagnosis of blistering lupus erythematous with a decrease in dermal sebaceous and eccrine glands. Direct immunofluorescence revealed deposits of IgG, complement C3, fibrinogen, IgM, and IgD in the epidermal corneal layer and the basement membrane zone. Reactivity to some dermal endothelial cell junctions and neural receptors was also noted. Also, complement C4 was positive against upper and middle dermal thin fibers. Complement D2 was positive around some enlarged dermal vessels, colocalizing with factor XIIIa. Our case highlights the complex and previously unreported immunologic response features associated with bullous lupus erythematosus.
Key words: Bullous lupus; IgD; Sebaceous glands; CD2
INTRODUCTION
As with systemic lupus erythematosus (SLE), bullous SLE has often been encountered in young adult females of African descent. In fact, bullous SLE may occur in all ages, sexes, and ethnicities, yet it tends to affect younger females [1,2]. Lupus nephritis has quite often been associated with bullous lupus, and this disease is rare in minors [3]. The lesions vary from blisters, to vesicles, to urticarial lesions, most often seen on the face or in other sun-exposed areas. In several cases, mucous membrane involvement has been reported. In general, skin biopsies with hematoxylin and eosin (H&E) staining show subepidermal detachment and dermal infiltration of neutrophils (often visualized near the epidermal-dermal junction) [1,2]. The clinicopathologic diagnostic criteria for bullous SLE include 1) a diagnosis of systemic lupus erythematosus (SLE) by the American College of Rheumatology (ARA) criteria; 2) a clinical blistering/vesicular eruption, and 3) histologic evidence of subepidermal blistering and a neutrophil-predominant dermal infiltrate. Oral and esophageal lesions have been described in bullous SLE [3]. Direct immunofluorescence (DIF) staining classically demonstrates linear and/or granular deposits of immunoglobulin IgG, IgM, and, frequently, IgA in the BMZ. A serrated pattern is not seen by DIF. These patterns assist in differentiating bullous lupus (with tissue-bound auto-antibodies against type VII collagen) from many other anti-BMZ disease antibodies (in which a non-serrated pattern is seen) [1]. Some reports have shown bullous SLE antibodies to bullous pemphigoid antigens (BP 230; BP 180) and laminins 5 or 6 with immunoblotting [4]; however, the supporting literature is not extensive enough to definitively correlate with the disease. Some researchers have attempted to differentiate types I and II of bullous SLE, yet the classification is not completely accepted. One proposal would subclassify bullous SLE as type I or II depending on whether the antigens are directed against type VII collagen (type I bullous SLE) versus against bullous pemphigoid antigens (BP 230, 180) or laminin 5 or 6 antigens (type II bullous SLE). Dapsone at a dose of 1.0–1.5 mg/kg/day is the treatment of choice. It is effective in most patients, leading to swift clinical improvement within days or several weeks [4]. In some cases in which dapsone is ineffective, prednisolone, rituximab, methotrexate, or azathioprine are utilized [4–6].
CASE REPORT
A 67-year-old black female with a forty-year history of systemic lupus erythematosus (SLE) diagnosed with the criteria by the American College of Rheumatology (ARA) presented to the dermatologist regarding the sudden presentation of blisters, dense vesicles, bullae, and erosions. These lesions were present both on normal and erythematous skin in sun-exposed sites (Fig. 1a). Skin biopsies of perilesional areas were taken and evaluated with H&E, as well as with immunohistochemistry (IHC) and direct immunofluorescence (DIF) as previously described [7]. A diagnosis of bullous SLE was established based on the clinical presentation and the histologic findings. H&E staining demonstrated a subepidermal blister with a heavy neutrophilic infiltrate in 1) the blister lumen; and 2) the upper dermis, mainly present in the papillary tips with additional fibrin and neutrophilic debris. Notably, sebaceous glands were decreased in the dermis. Dapsone at a dose of 1.0–1.5 mg/kg/day was administered and the lesions resolved.
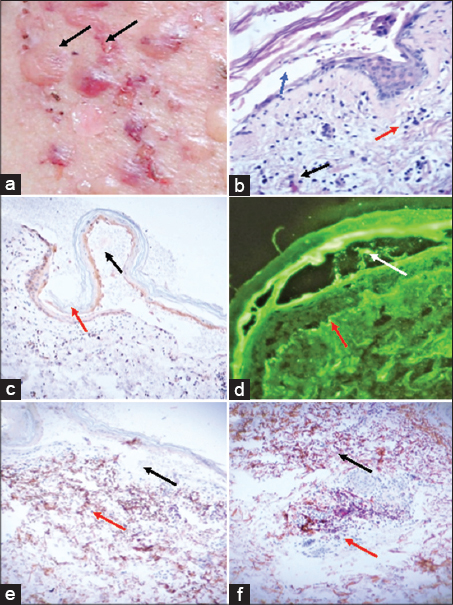 |
Figure 1: a: Clinical blisters and hemorrhagic blisters on the upper chest (black arrows). b: H&E staining displaying a partially re-epithelialized subepidermal blister (blue arrow) (200×). The black arrow highlights mucin in the dermis and the red arrow shows neutrophils around dermal blood vessels. c: IHC staining using IgD revealing a linear deposit along the base of the epidermis (brown staining; black arrow) as well as a lack of staining in the epidermal stratum spinosum (red arrow) (100×). d: DIF showing a positive blister lumen with positive staining with FITC conjugated anti-human IgM (white arrow), as well as positive non-serrated BMZ staining (red arrow) (400×). e: IHC staining with complement C4 showing positive staining in the extracellular matrix of the upper dermis (brown staining; black arrow), as well in the middle dermis (brown staining; red arrow) and positive staining elongated cells resembling the shape of fibroblasts (100×). f: IHC positive red staining on a cluster of cells using complement D2 (black arrow) (200×). The brown staining is dermal fiber structures staining for complement C4. |
To better study this case, we employed both single- and double-color IHC staining, performed with the Leica Bond MAX automated system (Buffalo Grove, Illinois, U.S.) with Novolink™ detection and Compact Polymer™ technology as previously described [7]. Specifically, for primary staining, we used Bond Max refined red detection DS9390, alkaline phosphatase linker polymer, and fast red chromogen (red staining). For secondary staining, we used bond polymer refined detection DS9800, horseradish peroxidase linker polymer, and DAB chromogen (brown staining). Positive and negative controls were consistently performed. The following antibodies were used for the IHC: mouse anti-human monoclonal antibodies 1) Clone F7.2.38, Complement C4 (C4) Cat. No. F0169, 2) IgD Cat. No IR517, 3) factor XIIIa, 4) CD2 (LFA-2) Cat No: PA0271, all from Leica/Novocastra. For DIF staining, we used antibodies and techniques as previously described [7]. It is generally accepted that bullous systemic lupus erythematosus is transient in most cases and usually reverts with no further flares, sometimes leaving hypo- or hyper-pigmentation. Our patient had no pigment alterations after recovery.
Fig. 1a shows multiple blisters some with hemorrhagic contents. Fig. 1b shows some mucin deposition in the dermis on H&E. The histologic features were representative of bullous SLE. IHC staining for IgD revealed linear staining along the basement membrane zone (BMZ) (Fig. 1c).
Using IHC, Complement C4 was positive in the extracellular matrix of the upper dermis as well in the middle and deep dermis, with positive individual elongated cells resembling the shape of fibroblasts (Fig. 1e). Complement D2 was positive in a cluster in the dermis around the remnants of a sebaceous gland and in some vascular bundles between the middle and deep dermis (Fig. 1f). Factor XIIIa was also positive in these areas (Fig. 1f).
DIF staining displayed the following results: kappa light chains (+++, epidermal stratum corneum); lambda light chains (+++, epidermal stratum corneum); IgG (++, patchy and serrated in the BMZ); (-); complement C1q (+/-, linear BMZ); complement C3 (++, dotted in the BMZ, dotted in the epidermal and the mesenchymal endothelial cell junctions); albumin (-) and fibrinogen (++++, serrated linear BMZ, corneal layer and dermal perivascular). IgM was similar in positivity to fibrinogen, yet additional cytoid bodies were observed in the BMZ (Fig. 1d). IgD was positive (++++) in a linear fashion in the BMZ as well as in multiple small cell junctions in the upper and middle dermis. Please note that IgD was observed using both IHC and DIF in a similar pattern to that seen with fibrinogen.
DISCUSSION
Bullous systemic lupus erythematosus is also termed bullous eruption of SLE and vesiculobullous SLE. The association between collagen VII and bullous SLE was found using autoantibodies detected by indirect immunofluorescence, as well by enzyme-linked immunosorbent assays (ELISA) against type VII collagen [8]. In the current case, we clearly demonstrate positive staining in the BMZ with IgD. We also describe non-classical complement findings in the BMZ of the blisters.
The most common pattern of bullous SLE autoreactivity using DIF displays linear or granular immunoglobulins (IgG, IgM, IgA, and/or complement C3) in the dermal-epidermal junction (DEJ) along the BMZ in perilesional skin biopsies [8]. In the current case, IgD was also observed using both IHC and DIF staining in a similar pattern to that seen with fibrinogen.
Since we serve as a reference laboratory for blistering diseases, we have noted that not only bullous lupus, yet also other autoimmune blistering diseases display positivity when using IgD [9]. We have observed similar patterns of positivity across multiple disorders with IgD, IgM, and fibrinogen. Recent studies have revealed that high expression of the IgD-B-cell receptor (IgD-BCR) may help physiologically autoreactive B cells endure in peripheral lymphoid tissues owing to unresponsiveness to self-antigens; and help their entry into germinal centers to “redeem” autoreactivity via somatic hypermutation [10]. In the current case, we speculate that IgD may be helping in the attack on the self-antigen, potentiating the autoimmune response.
We also observed complement activation not usually reported in bullous lupus. In this case, complement C4 was highly positive. Interestingly, in the pathogenesis of tissue inflammation and injury in SLE, complement C4 is also higher in patients with lupus nephritis [11].
We conclude that the roles of IgD, complement C4, and other inflammatory markers may indeed play critical roles in bullous SLE and that these markers might be included in the diagnostic and research panels employed to study autoimmune diseases. Furthermore, low complement C4 levels are frequently found in systemic lupus erythematosus; patient seric levels may also fluctuate, with low levels of both often seen in disease flare-ups. In our case, the patient displayed a normal seric acid titer (< 8.3 mg/dL; normal range < 10 mg/dL) [12]. Thus, we speculate that the autoimmune response to selected structures in the dermis (demonstrated by IHC staining) may contribute to a decrease in complement C4 levels in patients with bullous SLE.
ABBREVIATIONS
Hematoxylin and eosin (H&amp;E), immunohistochemistry (IHC), direct immunofluorescence (DIF), basement membrane zone (BMZ), epidermolysis bullous acquisita (EBA), systemic lupus erythematosus (SLE), American College of Rheumatology (ARA), fluorescein isothiocyanate (FITC), 4’,6-diamidino-2-phenylindole (DAPI), complement component 4 (C4).
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki. The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published, and due effort will be made to conceal their identity.
REFERENCES
1. de Risi-Pugliese T, Cohen Aubart F, Haroche J, Moguelet P, Grootenboer-Mignot S, Mathian A, et al. Clinical, histological, immunological presentations and outcomes of bullous systemic lupus erythematosus:10 new cases and a literature review of 118 cases. Semin Arthritis Rheum. 2018;48:83-9.
2. Montagnon CM, Lehman JS, Murrell DF, Camilleri MJ, Tolkachjov SN. Subepithelial autoimmune bullous dermatoses disease activity assessment and therapy. J Am Acad Dermatol. 202;5:S0190-9622(21)00477-1.
3. Tanaka H, Jimbo Y, Iwane K, Takata R, Ikeda A, Dougaki M, et al. [A case of bullous systemic lupus erythematosus with sloughing esophagitis]. Nihon Shokakibyo Gakkai Zasshi. 2020;117:252-60.
4. Odonwodo A, Vashisht P. Bullous systemic lupus erythematosus. StatPearls 2021 https://www.ncbi.nlm.nih.gov/books/NBK557445
5. Shi H, Gudjonsson JE, Kahlenberg JM. Treatment of cutaneous lupus erythematosus:Current approaches and future strategies. Curr Opin Rheumatol. 2020;32:208-14.
6. Duan L, Chen L, Zhong S, Wang Y, Huang Y, He Y, et al. Treatment of bullous systemic lupus erythematosus. J Immunol Res. 2015;2015:167064.
7. Abreu Velez AM, Upegui-Zapata YA, Valencia-Yepes CA, Upegui-Quiceno E, Mesa-Herrera NR, Jiménez-Echavarria AM, et al. Membrane attack complex (C5b-9 complex or Mac), is strongly present in lesional skin from patients with endemic pemphigus foliaceus in El Bagre, Colombia. J Cutan Pathol. 2019;46:925-9.
8. Sun S, Zhong B, Li W, Jin X, Yao Y, Wang J, et al. Immunological methods for the diagnosis of oral mucosal diseases. Br J Dermatol. 2019;181:23-36.
9. Abreu Velez AM, Smoller BR, Howard MS. An extraordinary case of oral leukocytoclastic vasculitis demonstrating IgD deposition around dermal vessels and within mucosal keratinocytes. Our Dermatol Online. 2021;12:50-3.
10. Wan Z, Zhao Y, Sun Y. Immunoglobulin D and its encoding genes:An updated review. Dev Comp Immunol. 2021;124:104198.
11. Weinstein A, Alexander RV, Zack DJ. A review of complement activation in SLE. Curr Rheumatol Rep. 2021;10;23:16.
12. Wu YL, Higgins GC, Rennebohm RM, Chung EW, Yang Y, Bi Zhou B, et al. Three distinct profiles of serum complement C4 proteins in pediatric systemic lupus erythematosus (SLE) patients:Tight associations of complement C4 and C3 protein levels in SLE but not in healthy subjects. Adv Exp Med Biol. 2006;586:227-47.
Notes
Source of Support: This work was supported by funding from Georgia Dermatopathology Associates, Atlanta, Georgia USA.,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-7692-4133 http://orcid.org/0000-0002-7301-8274 http://orcid.org/0000-0003-0430-6093 http://orcid.org/0000-0002-7692-4133 http://orcid.org/0000-0002-7301-8274 http://orcid.org/0000-0003-0430-6093 |

Comments are closed.