Advanced ulcerative tumefactive lesions of the mucocutaneous surface in previously unrecognized Langerhans Cell Histiocytosis
Fatima Azzahra Elgaitibi 1, Soumaya Hamich1, Noureddine Mahiou2, Kaoutar Znati3, Merieme Meziane1, Nadia Ismaili1, Leila Benzekri1, Karima Senouci1
1, Soumaya Hamich1, Noureddine Mahiou2, Kaoutar Znati3, Merieme Meziane1, Nadia Ismaili1, Leila Benzekri1, Karima Senouci1
1Department of Dermatology, Mohammed V university in Rabat, Ibn Sina University Hospital, Rabat, Morocco, ²Department of Otorhinolaryngology, Mohammed V University in Rabat, Ibn Sina University Hospital, Rabat, Morocco, 3Department of histopathology, Mohammed V University in Rabat, Ibn Sina University Hospital, Rabat, Morocco
Corresponding author: Dr. Fatima Azzahra Elgaitibi
How to cite this article: Elgaitibi FA, Hamich S, Mahiou N, Znati K, Meziane M, Ismaili N, Benzekri L, Senouci K. Advanced ulcerative tumefactive lesions of the mucocutaneous surface in previously unrecognized Langerhans Cell Histiocytosis. Our Dermatol Online. 2021;12(4):419-421.
Submission: 26.07.2020; Acceptance: 23.10.2020
DOI: 10.7241/ourd.20214.15
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Langerhans cell histiocytosis (LCH) is an uncommon systemic disease characterized by the infiltration of one organ or more by Langerhans cells. Its clinical presentation is heterogeneous and depends on the affected organs. We report the rare case of a 43-year-old female presenting herself with central diabetes insipidus and receiving nasal desmopressin spray. Later, the patient developed multiple papules and pustules progressing to multiple ulcers affecting the retroauricular and axillary folds, as well as the oral and genital mucosas. The skin lesions had been evolving for several months. A histopathological examination of a skin biopsy confirmed the clinical suspicion of LCH.
Key words: Langerhans cell; Diabetes insipidus; Ulcers
INTRODUCTION
Langerhans cell histiocytosis (LCH) is an uncommon systemic disease characterized by the infiltration of one organ or more by dendritic cells known as Langerhans cells [1]. It has an incidence rate of 3–5 cases per million [2].
Its clinical presentation is heterogeneous and affects preferentially the bone, skin, lymph nodes, and the central nervous system.
We report the case of a female with cutaneous and mucous membrane ulcers found to have systemic Langerhans cell histiocytosis lasting several years.
CASE REPORT
A 43-year-old female presenting herself with central diabetes insipidus had been receiving nasal desmopressin spray. An annually following by cerebral MRI was decided.
Five years after the first diabetes insipidus diagnosis, we received the patient in our department of dermatology. She reported a one-year history of multiple papules and pustules progressing in several days to multiple ulcers affecting the retroauricular and axillary folds, as well as the oral and genital mucosas. She described the ulcers as painful and gradually enlarging and suffered from the symptoms of fatigue, galactorrhea, and amenorrhea.
A clinical examination revealed a large retroauricular fistula complicating mucopurulent ulcers, not well-demarcated, with raised edges, with an erythematous background and a fibrinous base localized in the pinna. The right axillary fold and the oral and genital mucosas were affected by similar ulcers (Fig. 1).
 |
Figure 1: Multiple mucopurulent ulcers affecting the retroauricular folds with a large fistula, the axillary fold, and the genital mucosa. |
A histopathological examination of a skin biopsy specimen revealed a dense proliferation of histiocytes immunoreactive to PS100, CD1a, and langerin (Fig. 2).
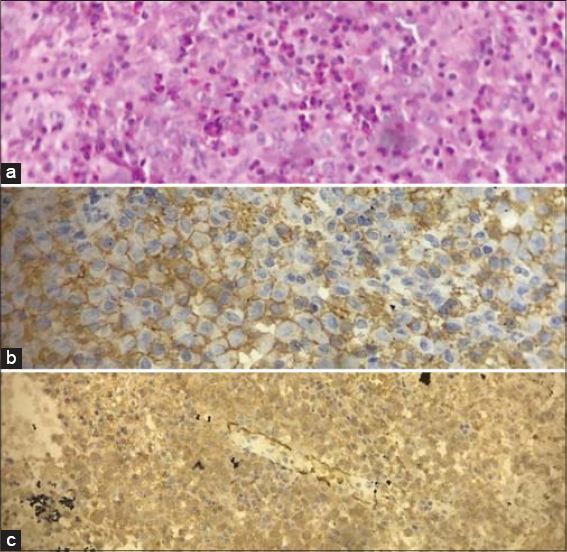 |
Figure 2: (a) A dense proliferation of histiocytes (H&E, 20×). (b) Immunoreactivity for CD1a (H&E, 20×). (c) Immunoreactivity for PS100 (H&E, 10×). |
A blood test confirmed the clinical suspicion of panpituitarism.
Gadolinium-enhanced orbito-cerebral MRI revealed a hypothalamic mass invading the optic chiasma (Fig. 3). Thoracic, abdominal, and pelvic CT revealed mediastinal and pulmonary parenchyma involvement. A skeletal X-ray examination was normal.
 |
Figure 3: Magnetic resonance imaging images with contrast enhancement: Enhanced lesion invading the optic chiasma. |
After the diagnosis of multisystemic LCH was confirmed and the extent of the disease evaluated, the patient received a combination of vinblastine and prednisolone as therapy.
DISCUSSION
LCH is characterized by the proliferation and accumulation of Langerhans cells in tissues, most often organized in granulomas. In 2016, the Histiocyte Society classified Langerhans cell histiocytosis as an inflammatory myeloid neoplasm [3].
Its clinical presentation depends on whether there is single- or multiple-organ involvement, which may lead to death, especially if the bone marrow, liver, spleen, and lungs are affected. Multisystemic disease is most frequently seen in younger children while unique lesions are more common in adults [4,5].
The most typical skin presentation is multiple erythematous, squamous, and crusted papules. Seborrheic or eczematiform lesions may also be part of the clinical syndrome. Cutaneous lesions most often affect the trunk, face, and scalp. External auditory canal and retro-auricular fold involvement are suggestive of LCH.
The central nervous system is frequently affected. Posterior pituitary involvement is common and may precede the diagnosis of LCH. The involvement of the anterior pituitary is also possible, sometimes leading to panhypopituitarism [2,6]. MRI may reveal loss of hyperintense signals in the posterior pituitary on T1-weighted images, as in any central DI signifying loss of ADH storage granules with contrast enhancement, a thickened pituitary stalk or, more rarely, a pituitary tumor that may press on nearby structures [7].
The bone is the most frequent location of LCH. It may be unique or multiple, symptomatic or not, preferably in the axial and cephalic skeleton. Radiology demonstrates multiple, punched out, osteolytic lesions without peripheral condensation [6].
A histological examination revealed infiltration by Langerhans cells that immunoreacted to CD1a and langerin.
The management of LCH has not been consensually defined due to its low incidence. Adults with a diagnosis of LCH must have an assessment of disease extension, which includes a hematologic, pulmonary, renal, hepatic, and skeletal systemic evaluation. Langerhans cell histiocytosis is subdivided into two types of involvement: single-organ with multifocal or unifocal involvement and multiple-organ with or without high-risk organ abnormality.
The treatment of LCH depends on the extent of the disease. Cutaneous LCH is generally treated with local therapies, whereas multisystemic LCH with multiple-organ involvement requires multi-drug therapy using, as in children, vinblastine coupled with prednisolone. The discovery of the role of the BRAF mutation as a key activator of Raf–MEK–ERK signaling in LCH opens the door to new therapeutic perspectives, especially targeted therapies [1].
The accessibility of a skin biopsy performed on the genital ulcer and the right axillary fold gave us a histopathological diagnosis of Langerhans cell histiocytosis. Given the fact that a biopsy was unattainable from the other sites, a specimen study of a shallow skin biopsy was the less invasive way to diagnose this uncommon and intricate disease. Moreover, it allowed us to reveal the etiology of the symptoms that began several years ago with diabetes insipidus before the appearance of the genital ulcers and other skin lesions.
CONCLUSION
The main characteristic observed in our patient was the clinical polymorphism of cutaneous manifestations. The atypical evolution of the papules and pustules into deep and mutilating ulcerous involvement was an uncommon skin presentation.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Néela A, Artifonia M, Donadieuc J, Lorillon G, Hamidou M, Tazi A. Langerhans cellhistiocytosis in adults. Rev Med Interne. 2015 Oct;36(10):658-67
2. Kobayashi M, Tojo A. Langerhans cell histiocytosis in adults:Advances in pathophysiology and treatment.Cancer Sci. 2018 Dec;109(12):3707-13.
3. Emile JF, Abla O, Fraitag S, Horne A, Haroche J, Donadieu J and al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016 Jun 2;127(22):2672-81.
4. Girschikofsky M, Arico M, Castillo D, Chu A, Doberauer C, Fichterand J and al. Management of adult patients with Langerhans cell histiocytosis:Recommendations from an expert panel on behalf of Euro Histio net.Orphanet J Rare Dis 2013;8:72.
5. Tamai S, Ueno M, Hayashi Y, Sasagawa Y, Watanabe T, Murakami K et al. Enlargement of Langerhans cell histiocytosis of the hypothalamus with progression into the basal ganglia and white matter. Surg Neurol Int. 2018;9:197.
6. El-Arab KK, Luedke AI, Julian BT, Ferrauiola J, Miller FR, Wang HT. Langerhans cell histiocytosis in an adult:a discussion of epidemiology and treatment options. J Craniofac Surg. 2020;31:e70-3.
7. Redhu R, Nadkarni T, and R. Mahesh. Diabetes insipidus associated with a thickened pituitary stalk in a case of Langerhans Cell Histiocytosis. J Pediatr Neurosci. 2011 Jan-Jun;6(1):62–64.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-0313-6152 http://orcid.org/0000-0002-0313-6152 |

Comments are closed.