BAP1 mutation in a patient with oculocutaneous albinism
Daniel Bax , Evan Piette, Prajesh Adhikhari
, Evan Piette, Prajesh Adhikhari
University of Vermont Department of Medicine, Division of Dermatology, Burlington, VT 05401, USA
Corresponding author: Daniel Bax, MD
How to cite this article: Bax D, Piette E, Adhikhari P. BAP1 mutation in a patient with oculocutaneous albinism. Our Dermatol Online. 2021;12(3):270-272.
Submission: 14.12.2020; Acceptance: 27.02.2021
DOI: 10.7241/ourd.20213.8
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Dermatologists and dermatopathologists are uniquely positioned to identify BRCA1-associated protein (BAP1)-related cutaneous diseases. Recognition of BAP1 mutations is critical to patients and their families to assure they are appropriately counseled and screened for malignancy. A twenty-year-old male with a history of oculocutaneous albinism type 2 (OCA2) and basal cell carcinoma (BCC) presented to the dermatologist for a full-body skin examination. A 7 mm erythematous papule on the left upper back was biopsied and found to be a BAP1-inactivated melanocytic tumor (BIMT). To our knowledge, this is the first case of oculocutaneous albinism of any type and a BAP1 germline mutation presenting in the same patient.
Key words: Albinism; BAP1; BAPoma; OCA2; Oculocutaneous
INTRODUCTION
The term albinism refers to a number of diseases unified by the genetic inheritance of reduced melanin production. Oculocutaneous albinism (OCA) encompasses a number of autosomal recessive inherited diseases in which melanin production is absent or reduced in various ectodermally derived tissues, including the eyes, skin, and hair [1]. OCA2 is the most prevalent type of OCA worldwide [2]. The reduction in melanin manifests itself in increased actinic damage and, subsequently, increased incidence of squamous cell carcinomas (SCCs), BCCs, and melanomas in these patients [3–5].
Mutations in the BAP1 tumor-suppressor gene are inherited in an autosomal dominant pattern and predispose families to the development of various cutaneous neoplasms, including BCC, BAP1-inactivated melanocytic tumor (BIMT), and cutaneous melanoma. Beyond cutaneous diseases, this mutation also puts patients at higher risk for internal malignancies, including uveal melanoma, renal cell carcinoma, and malignant mesothelioma [6,7]. Dermatologists and dermatopathologists are uniquely positioned to identify BAP1-associated cutaneous diseases. Recognition of BAP1 mutations is critical to patients and their families to assure they are appropriately counseled and screened early for malignancy. Herein, we present a case of oculocutaneous albinism type 2 and a BAP1 germline mutation in the same patient.
CASE REPORT
A twenty-year-old male with a history of oculocutaneous albinism type 2 and basal cell carcinoma (BCC) presented to the dermatologist for a full-body skin examination. The examination was notable for a depressed and elongated 4 mm papule on the left nasal bridge as well as a 7 mm erythematous papule on the upper left back (Fig. 1). The lesions were anesthetized and two shave biopsies were performed in the typical fashion.
 |
Figure 1: A 7 mm erythematous papule on the left upper back. |
Histopathology of the lesion on the left nasal bridge revealed BCC. Histopathology of the lesion on the upper left back revealed a predominantly intradermal, compound, melanocytic proliferation with nuclear pleomorphism, multinucleated cells, and rare mitoses (Fig. 2a). BAP1 immunostaining revealed no staining of the involved melanocytes (Fig. 2b). A diagnosis of a BAP1-inactivated melanocytic tumor (BIMT) was confirmed.
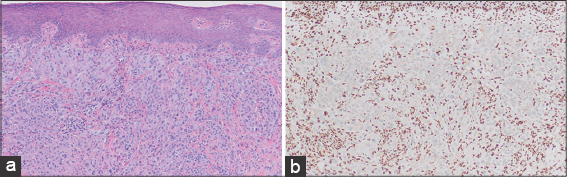 |
Figure 2: (a) Closely apposed nests of large, oval melanocytes with eccentric nuclei and admixed lymphocytes in the lesion on the upper back (H&E; 10×). (b) A notable lack of staining of spitzoid melanocytes comprising the bulk of the tumor on the upper back; positive staining of keratinocyte nuclei in the overlying epidermis, lymphocytes, as well as smaller adjacent melanocytes (BAP-1 immunostaining; 10×). |
Subsequent immunostaining of the patient’s BCC on the left nasal sidewall (Fig. 3a) revealed diminished staining for BAP1 (Fig. 3b), further supporting an inherited BAP1 germline mutation. Mohs surgery and a simple excision were performed for the BCC and BIMT, respectively. The patient was referred to an ophthalmologist and for a clinical genetics program at our institution.
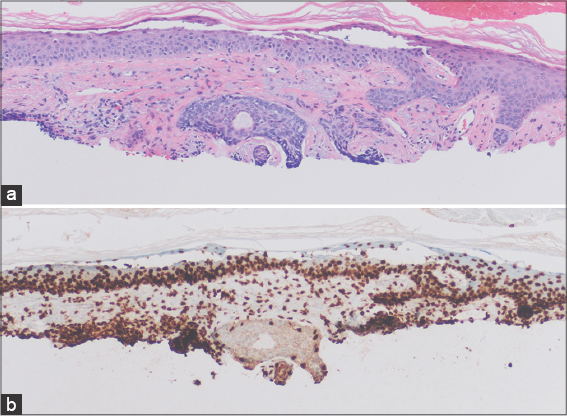 |
Figure 3: (a) Basal cell carcinoma on the nasal sidewall (H&E; 20×). (b) Diminished BAP-1 staining of the lesion on the nasal sidewall (BAP-1 immunostaining; 20×). |
DISCUSSION
Oculocutaneous albinism type 2 (OCA2) is an autosomal dominant disease caused by mutations in the OCA2 gene (formerly known as the P gene) on chromosome 15q11. The function of the P protein is yet to be outlined in detail but research suggests a likely role in regulating the pH of various organelles as well as facilitating glutathione accumulation within vacuoles [1]. Hypopigmentation seen in a small subset of patients with Prader–Willi syndrome (PWS) and Angelman syndrome (AS) represents a form of OCA2 as a result of contiguous imprinting deletions of chromosome 15q, which includes the OCA2 gene [8].
In the U.S., the overall prevalence of OCA2 is estimated at around 1:36,000, but is much higher in individuals of African descent [2]. The clinical spectrum of OCA2 is broad. Young patients show some pigmentary dilution of the hair, skin, and iris. Although pigmented melanocytic nevi and lentigines commonly develop in sun-exposed areas, patients have little to no ability to tan and, thus, tend to sustain a large burden of actinic damage over their lifetimes [1,3]. This actinic damage, and the subsequent cutaneous mutation burden, manifests itself in an increased incidence of skin cancers, including SCC, BCC, and melanoma in these patients [3–5].
BRCA1-associated protein (BAP1) is a gene on chromosome 3p encoding a deubiquitinating enzyme. This enzyme has been shown to play a role in multiple cellular processes, including cellular differentiation, transcription, regulation of the cell cycle, and DNA repair [9]. Mutations and deletions in BAP1 were first reported in association with breast and lung cancers. Wiesner and colleagues described a tumor predisposition syndrome involving BAP1 mutations inherited in an autosomal dominant pattern. Most notably, this syndrome includes the development of 5-50 skin-colored, red, and brown dome-shaped papules during the second decade of life [10].
The nomenclature of this neoplasm is still debated and terms to describe it include BAP1-inactivated melanocytic tumor (BIMT), melanocytic BAP1-mutated atypical intradermal tumor (MBAIT), atypical Spitz tumor (AST), a cutaneous atypical and epithelioid melanocytic lesion, BAPoma, a melanocytic nevus/tumor with a BAP1 mutation, and a Wiesner nevus.
Histopathology of the tumor is partially reminiscent of the Spitz nevus, but the characteristic features of Spitz nevi, including epidermal hyperplasia, Kamino bodies, clefting adjacent to nested melanocytes, hypergranulosis, and spindled melanocytes, are normally absent in BIMT [11]. It is challenging to reliably distinguish a BIMT from a spitzoid melanoma and research has demonstrated a significant lack of interobserver agreement, even among experts [12].
Continued studies on families with germline BAP1 mutations have further described this tumor predisposition syndrome. In addition to BIMTs, these patients have increased risk of uveal and cutaneous melanomas, mesothelioma, lung adenocarcinoma, clear cell renal cell carcinoma, and BCC [6,7].
Although commonly following a benign course, there is still insufficient data to predict the clinical behavior of BIMTs, and a simple lesion excision is usually recommended. More importantly, early recognition and diagnosis of BIMTs aids in establishing an underlying germline mutation in BAP1 and facilitates early screening and detection of malignant neoplasms in the patient and their family. An awareness of this clinical entity and the underlying BAP1 tumor predisposition syndrome uniquely positions dermatologists and dermatopathologists to identify this high-risk cohort of patients [13].
CONCLUSION
To our knowledge, this is the first case of oculocutaneous albinism of any type and a BAP1 germline mutation appearing in the same patient. This unfortunate coincidence of an increased UV-induced mutation burden (secondary to OCA2) and a tumor predisposition syndrome (germline mutation in BAP1) theoretically put our patient at an even greater risk of developing malignant cutaneous and ocular disease throughout his life. We are planning to continue with aggressive screening and close monitoring by both dermatology and ophthalmology. Our patient was adopted and, thus, family counseling was not necessary. Further studies are needed to determine if mutations in both the OCA2 and BAP1 genes are purely coincidental or if there are other factors increasing the probability for the co-occurrence of these mutations.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Raju BP, Nagaraju U, Raveendra L, Vivekananda, Sundar PK, Keshavalu L. Oculocutaneous albinism complicated with an ulcerated plaque. Our Dermatol Online. 2013;4:208-11.
2. Marçon CR, Maia M. Albinism:Epidemiology, genetics, cutaneous characterization, psychosocial factors. An Bras Dermatol. 2019;94:503-20.
3. Varma R, Asokan N, Sarin A, Rahman N. Unusually localised cutaneous multifocal squamous cell carcinoma. Our Dermatol Online. 2019;10:170-2.
4. Lekalakala PT, Khammissa RA, Kramer B, Ayo-Yusuf OA, Lemmer J, Feller L. Oculocutaneous albinism and squamous cell carcinoma of the skin of the head and neck in Sub-Saharan Africa. J Skin Cancer. 2015;2015:167847.
5. Mabula JB, Chalya PL, Mchembe MD, Jaka H, Giiti G, Rambau P, Masalu N, Kamugisha E, Robert S, Gilyoma JM. Skin cancers among Albinos at a University teaching hospital in Northwestern Tanzania:A retrospective review of 64 cases. BMC Dermatol. 2012;12:5.
6. Haugh AM, Njauw CN, Bubley JA, VerzìAE, Zhang B, Kudalkar E. Genotypic and phenotypic features of BAP1 cancer syndrome:A report of 8 new families and review of cases in the literature. JAMA Dermatol. 2017;153:999-1006.
7. Mochel MC, Piris A, Nose V, Hoang MP. Loss of BAP1 expression in basal cell carcinomas in patients with germline BAP1 mutations. Am J Clin Pathol. 2015;143:901-4.
8. Bolognia J, Jorizzo J, Schaffer J. Dermatology. Fourth ed. Elsevier Saunders;2018.
9. Wang A, Papneja A, Hyrcza M, Al-Habeeb A, Ghazarian D. Gene of the month:BAP1. J Clin Pathol. 2016;69:750-3.
10. Wiesner T, Obenauf AC, Murali R, Fried I, Griewank KG, Ulz P, et al. Germline mutations in BAP1 predispose to melanocytic tumors. Nat Genet. 2011;43:1018-21.
11. Wiesner T, Kutzner H, Cerroni L, Mihm MC Jr, Busam KJ, Murali R. Genomic aberrations in spitzoid melanocytic tumours and their implications for diagnosis, prognosis and therapy. Pathology. 2016;48:113-31.
12. Wiesner T, Murali R, Fried I, Cerroni L, Busam K, Kutzner H, et al. A distinct subset of atypical Spitz tumors is characterized by BRAF mutation and loss of BAP1 expression. Am J Surg Pathol. 2012;36:818-30.
13. Zhang AJ, Rush PS, Tsao H, Duncan LM. BRCA1-associated protein (BAP1)-inactivated melanocytic tumors. J Cutan Pathol. 2019;46:965-72.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
|

Comments are closed.