Psoriasis, pityriasis alba, and vitiligo (PPV) are a triad of one disease: New observation
Khalifa E. Sharquie1,2, Inas K. Sharquie3, Ali N. Al Hamza 2,4
2,4
1Department of Dermatology, College of Medicine, University of Baghdad, Iraq, 2Deprtment of Dermatology, Medical City Teaching Hospital, Baghdad, Iraq, 3Department of Microbiology & Immunology, College of Medicine, University of Baghdad, Baghdad, Iraq, 4College of Medicine/ University of Basrah, Iraq
Corresponding author: Ali N. Al Hamza, MBChB, MS
How to cite this article: Sharquie KE, Sharquie IK, Al Hamza AN. Psoriasis, pityriasis alba, and vitiligo (PPV) are a triad of one disease: New observation. Our Dermatol Online. 2021;12(3):314-323.
Submission: 02.08.2020; Acceptance: 01.11.2020
DOI: 10.7241/ourd.20213.21
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Keratinocytes and melanocytes live in the epidermis, which is considered to be a very good environment for both. Therefore, any change in this regard will affect the function of both of them. Keratinocytes form the main bulk of the epidermis and are the only melanin storage as the dendrites of the basal melanocytes run between keratinocytes and gift melanin granules to surrounding keratinocytes. Consequently, any immunoinflammatory reaction in the epidermis will influence the function of both of them. Recent evidence has shown that psoriasis, pityriasis alba, and vitiligo are closely related diseases and might reflect one etiopathogenesis initially targeting the hair follicles and then extending into the epidermis proper. Hence, the three diseases form one condition: the so-called PPV triad. Consequently, the objective of the following work is to show how these three skin problems are closely related clinically, immunologically, and pathologically, thus constituting the PPV triad.
Key words: Psoriasis; Pityriasis alba; Vitiligo; PPV Sharquie triad; Follicular leukoderma
EPIDEMIOLOGY AND PATHOPHYSIOLOGY OF PSORIASIS
Psoriasis is a common chronic inflammatory skin disease affecting 1–3% of the population [1]. Psoriasis vulgaris, the most common variant, characterized by scaly erythematous plaques, mainly affects the epidermis and involves the extensor surfaces, although any cutaneous site may be affected. Follicular psoriasis, although common, is not well recognized in the dermatological literature [2]. This could be explained by the fact that the initial event in the clinical picture of psoriasis is minute or even microscopic, a transient lesion affecting the upper stable segment of hair follicles and progressing into ordinary psoriasis (Figs. 1 and 2). Psoriasis is a multifactorial disease with a complex pathogenesis. Normally, the skin epidermis shows regular and constant turnover, occurring every 26–28 days [3]. In comparison to a normal skin epidermis, a psoriatic epidermis manifests itself with hyperplasia, rapid turnover, and intensified mitosis [4]. Psoriatic skin turnover occurs every 3–4 days [5] leading to keratinocyte hyperproliferation, which is a characteristic feature of psoriasis. Several cell types and cell–cell interactions have been involved in the disease process, including keratinocytes, antigen-presenting cells, Langerhans cells, T cells, macrophages, and natural killer cells. In addition, various Th1 cytokines, keratinocyte growth factor (KGF), vascular endothelial growth factor (VEGF), and interleukins are suggested to play a role in psoriasis pathogenesis [6].
 |
Figure 1: A 25-year-old male patient with follicular psoriasis involving the trunk. |
 |
Figure 2: A 23-year-old male patient with follicular plaques of psoriasis involving the back. |
On the other hand, the genetic role is considered from the observation of a high concordance rate in monozygotic twins (63–73%) compared to a lower rate in dizygotic twins (17–20%). There is considerable heterogeneity in psoriasis, and different genetic loci have been identified (PSORS1–10) [7,8], although psoriasis susceptibility locus 1 (PSORS1) remains the most determinant genetic factor, accounting for 50% of genetic variance in psoriasis [9,10].
THE ROLE OF KERATINOCYTES IN PSORIASIS ETIOPATHOGENESIS
The exact sequence of events in the initiation of psoriasis remains unknown. Although activated T lymphocytes and IL-17 are crucial to the development and persistence of psoriatic lesions. The pathophysiology of psoriasis cannot be explained merely by T lymphocytes as other skin-resident cells, such as keratinocytes and dendritic cells, are also involved [11]. The epidermal skin plays a significant role in early skin lesions by activation and recruitment of immune and inflammatory cells. The psoriatic auto-antigen loop concept is considered in the pathogenesis of psoriasis [11]. Antimicrobial peptide LL37 (cathelicidin) is overexpressed in psoriatic skin and triggers the activation of innate immune cells. It has been found that 2/3 of moderate-to-severe psoriatic patients have LL-37–specific CD4 and/or CD8 T lymphocytes, which can produce INF-? and Th-17 cytokines. The presence of LL-37–specific T cells in circulation correlates significantly with the disease process [12]. ADAMTSL5 (A disintegrin and metalloprotease domain containing thrombospondin type 1 motif-like 5), which is a melanocyte-derived protein, has also been observed to be strongly expressed in keratinocytes throughout the epidermis along with scattered expression in some dermal blood vessels and other perivascular dermal cells in psoriasis [13]. This protein has recently been implicated as an activating antigen for IL-17–producing T cells in psoriasis [14]. Both LL-37 and ADAMTSL5 autoantigens are downregulated following treatment with etanercept and IL-17 blockade [15]. Lande et al. identified the role of LL-37 in breaking innate tolerance to self-DNA as a fundamental player in the autoimmunological process of psoriasis. They found that LL-37 converts self-DNA into a potent trigger for INF production by forming aggregates and condensed structures that stimulate plasmacytoid dendritic cells (pDCs) through toll-like receptor 9 TLR9 [16,17]. Also, LL-37 binds to self-RNA and activates myeloid dendritic cells (mDC) through TLR7 and TLR8 in psoriatic lesions, leading to IL-6 and TNF production and differentiation of mDCs into mature DCs [18].
Furthermore, as keratinocyte autocrine stimulation by IL-1 exists in the epidermis, an experimental aberration of this highly controlled autocrine function has been tested using double transgenic mice, which show overexpression of the functional IL-1 receptor and 17 kD IL-1a in basal keratinocytes. It has been found that IL-1 has the ability to cross the basement membrane and activate dermal cells in addition to activating nearby cells such as keratinocytes, Langerhans cells, and melanocytes. Also, it leads to a wide variety of secondary responses, including the induction of secondary cytokines such as IL-6 and GM-CSF, leading to epidermal hyperplasia and dermal inflammatory cell infiltrate [19].
The characteristic histopathological findings of psoriasis include uniform elongation of the rete ridges, thinning of the suprapapillary plate, dilated blood vessels, intermittent parakeratosis, neutrophil aggregates in the epidermis, and perivascular infiltration of lymphocytes [20].
VITILIGO: EPIDEMIOLOGY AND FOLLICULAR LEUKODERMA
Vitiligo is an acquired depigmenting skin disorder affecting approximately 1% of the population worldwide, and is considered one of the most common dermatoses [21]. Generally, two major types of vitiligo are categorized clinically. The more common type is the nonsegmental, or generalized, variant, which has a gradual onset of symmetrical and nondermatomal distribution. The less common variant is the segmental vitiligo, which has a rapid onset and unilateral semidermatomal distribution and stabilizes once fully developed [22]. Follicular vitiligo is not an uncommon variant, commonly seen in segmental and nonsegmental vitiligo [23]. Sharquie et al. observed that all types of vitiligo begin in the upper stable segment of hair follicles, and the initial lesions, similarly to psoriasis, may be minute or even microscopic and transient, leading to follicular leukoderma and progressing into ordinary vitiligo (Fig. 3). In the case of graying of hair, the loss of melanocytes is from the hair matrix, while in ordinary vitiligo, the loss of melanocytes is from the infundibulum and the basal layer of the epidermis. Another uncommon subtype is mucosal vitiligo, with a restricted involvement of the genital or oral mucosa [24].
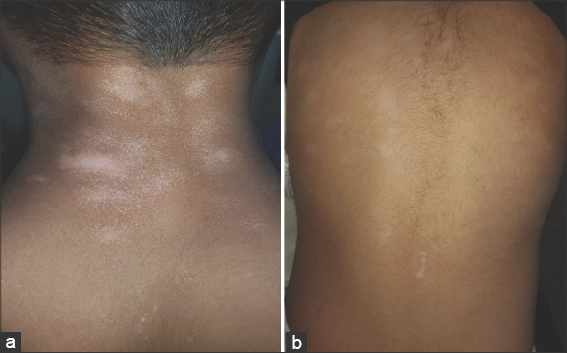 |
Figure 3: (a) A 25-year-old and (b) a 23-year-old patient with follicular vitiligo involving the back, showing follicular leukoderma, some coalescing to form patches of ordinary vitiligo. |
Stages of Depigmentation in Vitiligo
Vitiliginous skin lesions usually progress in two stages of depigmentation. In stage one, the skin lesion is whitish-brown with a partial pigmentation loss and stays for weeks to months. In stage two, it turns ivory white and loses all pigmentation (Fig. 4). In both stage one and two, marginal and even normal uninvolved skin shows lymphocytic epidermal cell infiltrates, which are significantly higher than the normal skin control and are markedly higher in stage-one vitiligo specimens. This staging of pigmentation loss may be observed affecting the scalp hair as, in stage one, the hair appears blonde because of the partial melanin loss (Fig. 5).
 |
Figure 4: A 7-year-old male with vitiligo showing stage-one partial pigmentation loss progressing into stage-two complete pigmentation loss, with small ivory-white macules. |
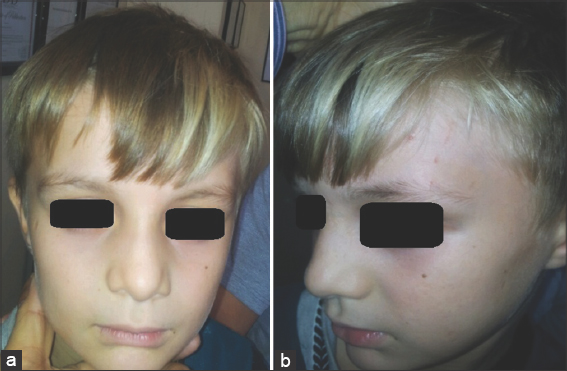 |
Figure 5: (a-b) An 8-year-old male patient with vitiligo showing stage-one pigmentation loss affecting the scalp hair, which changed into whitish-brown blonde. |
Etiology of Vitiligo
The etiology of vitiligo is complex and the available data supports the development of autoimmune phenomena in genetically predisposed individuals [25]. Several theories have emerged to explain the pathogenesis of vitiligo, mainly including autoimmune, autodestructive, and neurohormonal theories [26–28]. Other studies have suggested a convergence theory, which claims that, in addition to the aforementioned theories, melanocytorrhagy, impaired melanocyte migration, and an altered cellular environment each contribute to the disease process and none is mutually exclusive [29]. A histopathological study of vitiligo lesions showed a significant decrease in or the complete absence of melanocytes and the presence of inflammatory cell infiltrates in vitiliginous skin, including the epidermis. Additionally, the inflammation may sometimes be more intense and severe in the epidermis, even forming Pautreier-like microabscesses with little inflammation in the dermis. This inflammatory reaction is more intense in stage-one pigmentation loss and in the marginal area. Other features, such as hyperkeratosis, acanthosis, spongiosis with exocytosis, melanophages, rete ridge elongations, and telangiectasia, may also be observed. Accordingly, vitiligo is considered an inflammatory skin disease and the epidermal lymphocytic infiltrate is most likely the primary immunological event [30]. This inflammatory reaction might begin in the hair follicles and then progress into the adjacent epidermis. This inflammatory reaction might be reflected clinically as an advancing erythematous ring at the peripheral border of the vitiligo area (Fig. 6).
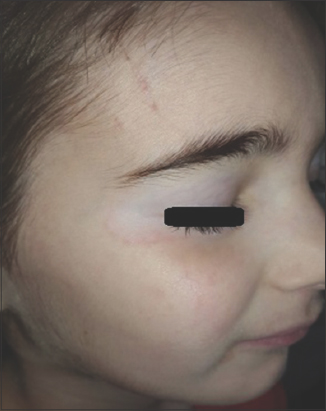 |
Figure 6: A 9-year-old female patient with vitiligo showing an advancing erythematous border of a vitiligo patch on the face. |
The Role of Keratinocytes in Vitiligo Etiopathogenesis
Epidermal homeostasis is critical for organism survival and any changes or defects in skin barrier function may predispose to cutaneous inflammation [31]. Most studies concern themselves with the abnormalities of melanocytes rather than keratinocytes. In fact, the melanocytes in the epidermis form functional units with the adjacent keratinocytes [32], as each melanocyte in the basal layer serves multiple surrounding keratinocytes. In vitro, cell-to-cell interaction and cross talk are observed in the differentiation and proliferation processes of melanocytes [33]. Keratinocytes produce several growth factors as well as cytokines that affect melanocytes function, proliferation, and differentiation processes [32]. For instance, endothelin 1, stem cell factor, and GM-CSF (granulocytes–monocytes colony stimulating factor) stimulate melanocyte proliferation and melanogenesis, while IL-6 cytokines and tumor necrosis factor alpha secreted by adjacent keratinocytes inhibit melanocyte function [26,34,35].
A recent study demonstrated an associated role of keratinocytes in the pathogenesis of vitiligo by measuring the keratinocyte expression level of liver X receptor alpha (LXR-a), a member of the nuclear hormone receptors that acts as a transcription factor. It has been found that LXR-a was upregulated in perilesional and lesional skin [31]. Numerous genes that regulate melanocyte function are governed by LXR-a. Activation of LXR-a by TO901317, a synthetic LXR ligand, inhibits melanogenesis through accelerated degradation of microphthalmia-associated transcription factor (MITF), a master transcriptional regulator of melanogenesis, by extracellular signal-regulated kinase (ERK) [36]. Reduction in the expression of KIT protein and its downstream effectors, including MITF-M, by melanocytes has also been detected by another study in lesional and perilesional sites of vitiligo [34]. Furthermore, structural keratinocyte abnormalities have been found to affect the growth and survival of melanocytes in vivo. E-cadherin, which mediates the adhesion between keratinocytes and melanocytes, has been shown to be absent or be in a discontinuous distribution across the melanocyte membrane in vitiligo patients. This defect is associated with the detachment of melanocytes from both basal and suprabasal epidermal layers [37]. Consequently, it leads to passive death of melanocytes due to loss of cell-to-cell contact and decrease in keratinocytes-derived growth factors [32]. These observations may explain the Koebner phenomenon in vitiligo under stressful skin conditions, such as trauma.
Psoriasis and Vitiligo as Close Relatives
Both psoriasis and vitiligo are inflammatory autoimmune skin diseases in which T lymphocytes play a role in their pathogenesis, and immune modulators, including topical corticosteroid, have been used successfully in their management [28,38]. A meta-analysis study conducted on a total of 120,866 psoriasis and 79,907 vitiligo patients found significantly increased odds for psoriasis in vitiligo patients, and vice versa [39]. In a study conducted on 1000 subjects, Sharquie et al. reported that the frequency of psoriasis among vitiligo patients was 6%, while 2% of psoriatic patients had vitiligo. Also, the frequency of reported family history of psoriasis among vitiligo patients and vice versa was 9.2% and 9.6%, respectively. In addition, it was found that psoriatic lesions were superimposed on vitiligo patches, and some lesions, especially in follicular psoriasis, had progressed into vitiligo (Figs. 7–10). This work suggested a close relative association between vitiligo and psoriasis [40].
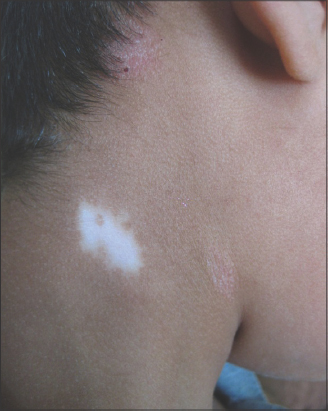 |
Figure 7: A 10-year-old male patient who developed a vitiligo lesion confined to a previously psoriatic lesion involving the right side of the neck. |
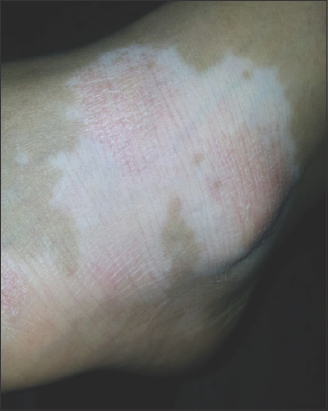 |
Figure 8: A 30-year-old female patient showing a coexistence of vitiligous and psoriatic lesions involving the left foot. |
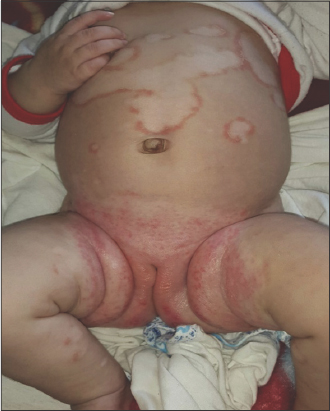 |
Figure 9: A one-year-old female patient with a coexistence of vitiligous and psoriatic lesions involving the napkin area and trunk. |
 |
Figure 10: An 8-year-old male patient showing (a) follicular psoriasis of the thigh and (b) segmental vitiligo of the scalp. |
Several case reports of vitiligo and psoriasis association have been mentioned in the literature. A case of vitiligo was observed in a patient who had guttate psoriasis and a positive family history of psoriasis with no other history of associated autoimmune diseases [41]. Menter et al. reported a case that developed multiple guttate psoriasis restricted totally to the areas of vitiligo lesions, suggesting that this pattern of anatomical cohabitation is sufficiently specific to make any coincidence unlikely [42]. Other cases of psoriatic lesions colonized with vitiligous patches have also been reported by other studies [43,44]. Such localization may be explained in several ways. Data from two research groups suggested the role of cytotoxic neurochemical mediators, such as substance P, as a possible mechanism of neuroecutaneous and dermatomal localization of affected areas in psoriasis and vitiligo [45,46]. In addition, the appearance of these two diseases may be due to expression of common cell surface markers that produces vitiligo as a result of autoimmune destruction and psoriasis as a result of autoimmune cellular stimulation through releasing of eicosanoids and lymphokine by keratinocytes [42]. Both diseases are affected by the pathogenic role of a high level of TNF-a [35,47]. In the case of vitiligo, TNF-a, IL-1, and IL-6 are powerful inducers of ICAM-1 in vitiligo and normal skin, which may enhance the target recognition of melanocytes by T lymphocytes, mediating cytotoxic damage [48]. The RNA released from necrotic keratinocytes might also act as an endogenous TLR3 ligand for the stimulation of ICAM-1 expression in human melanocytes [49]. TNF-a inhibits tyrosinase, tyrosine-related peptide 1 (TRP-1), and melanocyte proliferation in a dose-dependant manner [35,50]. In psoriasis, the elevated TNF-a level has been found in lesional skin and has been correlated with increased inflammation, which has successfully responded to TNF inhibitors [51]. Others have reported that this confined anatomical coexistence is possibly caused by the Koebner phenomenon [44]. Another explanation suggests the effect of cellular keratinocyte degeneration, which has been found in vitilignous skin [52]. This abnormality of keratinocytes may affect the uptake of melanosomes from adjoining melanocytes, which may lead to the accumulation of toxic intermediate metabolites in the melanin synthesis pathway. Hence, unchecked or aberrant melanin synthesis leads to cytotoxicity and destruction in a self-destruction mechanism not only to the melanocytes themselves but to the adjoining keratinocytes as well [52]. Melanocytes under stress undergo apoptosis, which also mediates the targeting of melanocytes by the immune system. If the immune response is sustained, melanocyte death may continue, leading to the spread of lesions to areas distant from the initial site [53]. Lastly, considering a genetic role, both diseases have been found to be linked by the same single-nucleotide polymorphism (SNP), rs9468925, at the HLA-C/HLA-B locus outside PSOR1 in the Han Chinese population [54].
Pityriasis Alba
Pityriasis alba is a common localized hypopigmenting disorder with numerous clinical variants [55], progressing in three stages. The first stage, or the early stage, begins as an erythematous patch with an elevated border lasting for several weeks. The second stage, or the intermediate stage, is characterized by the replacement of the patch with a smooth scaly layer (Fig. 11). Both of these two stages are marked by the presence of pinpoint follicular papules. Lastly, the third stage presents itself as a round hypopigmented macule with well-defined borders and loosely adherent scales [55]. This benign skin disorder predominantly affects children and adolescents between 3–16 years of age at a 5% prevalence rate [56]. It mainly affects the face, especially the cheeks. However, in 20% of affected children, the neck, arms, and shoulders are involved as well as the face. Less commonly, scattered lesions involve the trunk and limbs, sparing the face. Individual lesions are rounded, oval, or irregular hypopigmented patches that are usually not well marginated [57]. As pityriasis alba often coexists with atopy, it is considered a milder form of the disease [58]. However, pityriasis alba is not an entity by itself as it may be a manifestation of numerous other skin diseases, such as psoriasis, vitiligo, and other drying skin conditions, even as part of a tinea skin infection.
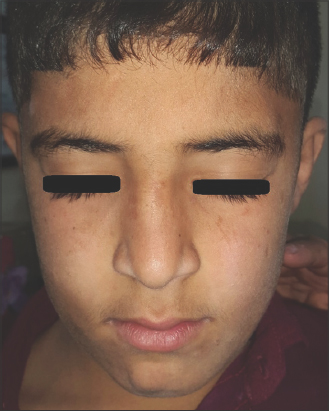 |
Figure 11: A 10-year-old patient with pityriasis alba showing scaly whitish erythematous patches with superimposed minute follicular papules involving the face. |
The pathogenesis of pityriasis alba remains a controversy. Some factors are reported to be associated with its development, including male sex, darker skin, exposure to sunlight, and atopy diathesis [59]. Other infectious causal factors, as microorganisms such as S. aureus, Pityrosporum, and Aspergillus, have been suggested but none has been confirmed [60]. However, in a study conducted in India and involving 200 patients, helminthic infestation and iron deficiency anemia were detected in 15.5% and 16.5% of patients, respectively [61]. However, these findings might be coincidental as malnutrition is a common issue among the Indian population. A histopathological study involving 56 patients found a marked reduction of melanin with no significant differences in the number of melanocytes between lesional and normal skin. However, ultrastructural observations revealed a reduction in melanosomes in the keratinocytes and degenerative changes in the melanocytes. Other histopathological features of pityriasis alba were found to be spongiosis with exocytosis, hyperkeratosis, and acanthosis in decreasing order of frequency [60], and this histological picture may mimic that of early psoriasis and vitiligo [30].
Vitiligo and Pityriasis Alba
Both vitiligo and pityriasis alba are inflammatory hypopigmented skin diseases [62,63]. However, pigment loss differences can be observed. In pityriasis alba, it is usually partial, with a centrifugal extension beginning from the center of the lesion, and this partial pigmentation loss may be considered stage-one melanin loss as in vitiligo, while pigmentation loss in vitiligo is either partial (stage one) or complete (stage two) with well- or ill-defined demarcation from the surrounding skin. It is worth noting that both conditions may begin as a scaly erythematous dermatitis-like presentation and mimic each other [30,62,64].
In a study involving 134 patients with pityriasis alba, Sharquie et al. observed different variants: scaly hypopigmented patches in 44%, smooth hypopigmented patches in around 30%, and a combination of different stages in around 9%. Also, a coexistence of both pityriasis alba and vitiligo (Figs. 12 and 13) with a positive Wood’s lamp test was observed in 14% of patients. Up to 57% of these patients with combined lesions had a positive family history of vitiligo. Furthermore, 14 (43.75%) patients out of 32 with pityriasis alba showed progression into vitiligo patches. Nine biopsies showed complete absence of melanin in lesional skin in Fontana–Masson stain [64]. Accordingly, there are 4 findings that may explain the possible association of pityriasis alba with vitiligo, including a positive family history and a possible genetic link, the development of both skin lesions in the same patient, a positive Koebner phenomenon in pityriasis alba, and lastly progression of pityriasis alba in the same patient into vitiligo during a follow-up.
 |
Figure 12: Coexistence of pityriasis alba and vitiligo in (a) a 9-year-old female and (b) an 11-year-old male. |
 |
Figure 13: An 8-year-old male patient with pityriasis alba involving the face and vitiligo involving the scalp hair. |
Pityriasis Alba as a Manifestation of Psoriasis or Vitiligo
Hence, through clinical observation and research studies, we have found that vitiligo or psoriasis may present themselves in their early stages as pityriasis alba on the face (Fig. 14), and there have been several pityriasis alba cases coexistent with vitiligo lesions. Furthermore, some cases of pityriasis alba progressed, with time, into vitiligo [64] or psoriasis. In the same context, cases of pityriasis alba involving a lower limb in psoriasis distribution sites have also been reported [65]. In addition, a presentation of a follicular lesion involving the upper stable segment of hair follicles in the early stages of pityriasis alba, psoriasis, and vitiligo has been observed [2,23,66] along with the sharing of the same inflammatory cell infiltrates. All these findings suggest a shared pathology and an interchangeable behavior of these skin disorders.
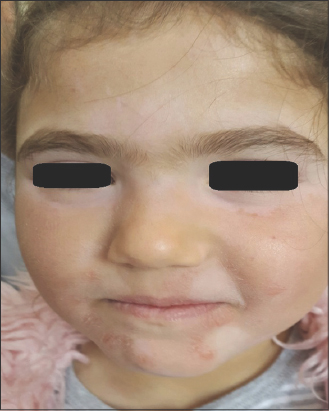 |
Figure 14: A 7-year-old female patient showing a coexistence of psoriasis, pityriasis alba, vitiligo lesions involving the face. |
Hair Follicles as the Early Target Area for Psoriasis, Pityriasis Alba, and Vitiligo
Suspended inside the dermis, hair follicles have a more extensive surface area that is more exposed to surrounding pathological damaging processes. In addition, the hair follicle has its separate blood supply. This does indicate a more intense inflammatory reaction in the hair follicle in comparison with the neighboring basal layer of the epidermis, where there is only one surface for basal keratinocytes that is exposed to the dermis. Also, antigenic stimulation may be more intense in hair follicles than on the epidermis, as the density of the various antigens and receptors may be increased in the keratinocytes of the outer root sheath of hair follicles than those of the epidermis. Accordingly, the immunological and pathological reaction should be more florid in the hair follicles [23,67,68]. Hence, we expect that all inflammatory skin diseases where the epidermis is the target area may begin at the hair follicles and may be more intense than the surrounding epidermal cells. This inflammatory reaction will show as minute macules or papules that coalesce together to form plaques or patches, as seen in vitiligo, psoriasis, seborrheic dermatitis, and many other diseases [58,67–70]. The immunological and inflammatory damage may involve the bulb of the hair, as in alopecia areata and graying of hair, or involve the infundibulum and isthmus, as in lichen planopilaris and discoid lupus erythematosus. Similarly, with hyperkeratosis of the skin, it will be more obvious in the orifices of hair follicles than in the surrounding epidermis, as seen in many cases of keratosis pilaris [68]. Although the diseases begin as follicular lesions, this change may be microscopic and difficult to see with the naked eye, transient, or clinically obvious and easily seen.
The question, thus, should arise as to why follicular lesions cannot always be observed clinically or histopathologically in these diseases. The following points aim to offer answers:
- The lesion might be so minute that it cannot be seen easily.
- The pathological changes might be so minimal that they cannot be observed on a histopathological section.
- The hair follicle involvement might have been missed during the sectioning of the biopsy.
- The follicular lesion might be transient and then spread to the actual epidermis.
Finally, what about the areas with no hair follicles, such as the palms, lips, and mucosas? In this case, the orifices of the sweat and salivary glands will represent the orifices of the hair follicles [71].
Hence, for the above reasons, the prevalence of actual follicular vitiligo, follicular psoriasis, and even follicular pityriasis alba cannot be well determined in the general population.
CONCLUSION
According to our clinical findings and research studies, in addition to a literature review, we have observed a close link between psoriasis, pityriasis alba, and vitiligo, wheras patients with pityriasis alba or psoriasis often develop vitiligo, and vice versa, or have a family history of psoriasis or vitiligo in favor of a common genetic background. Hence, this body of data supports our observation that psoriasis, pityriasis alba, and vitiligo constitute one triad on the basis of clinical, immunological, and histopathological findings (Fig. 15). Also, all these three diseases may begin in hair follicles and then progress to involve the epidermis. However, this may not be observed as the lesions might be transient, minute, or microscopic.
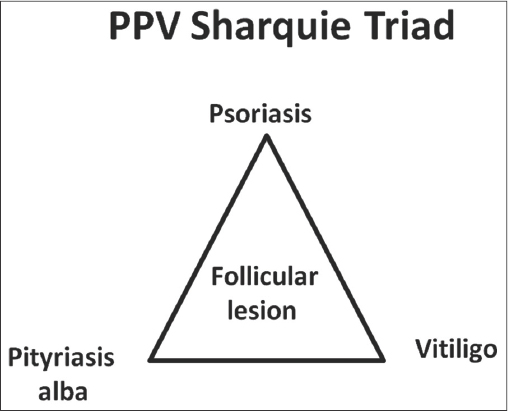 |
Figure 15: PPV Sharquie triad. |
REFERENCES
1. Langley RGB, Krueger GG, Griffiths CEM. Psoriasis:epidemiology, clinical features, and quality of life. Ann Rheum Dis. 2005;64 Suppl 2:ii18-23;discussion ii24-5.
2. Souza BCE, Bandeira LG, Cunha T, Valente NYS. Follicular psoriasis:an underdiagnosed entity?An Bras Dermatol. 2019;94:116-8.
3. Sotiropoulou PA, Blanpain C. Development and homeostasis of the skin epidermis. Cold Spring Harb Perspect Biol. 2012;4:a008383.
4. Weinstein GD, Van Scott EJ. Autoradiographic analysis of turnover times of normal and psoriatic epidermis. J Invest Dermatol. 1965;45:257-62.
5. Rothberg S, Crounse RG, Lee JL. Glycine-C-14-incorporation into the proteins of normal stratum corneum and the abnormal straum corneum of psoriasis. J Invest Dermatol. 1961;37:497-505.
6. Das RP, Jain AK, Ramesh V. Current concepts in the pathogenesis of psoriasis. Indian J Dermatol. 2009;54:7-12.
7. Ortonne JP. Recent developments in the understanding of the pathogenesis of psoriasis. Br J Dermatol. 1999;140 Suppl 54:1-7.
8. Chandran V. Genetics of psoriasis and psoriatic arthritis. Indian J Dermatol. 2010;55:151-6.
9. Jenisch S, Henseler T, Nair RP, Guo SW, Westphal E, Stuart P, et al. Linkage analysis of human leukocyte antigen (HLA) markers in familial psoriasis:strong disequilibrium effects provide evidence for a major determinant in the HLA-B/-C region. Am J Hum Genet. 1998;63:191-9.
10. Clop A, Bertoni A, Spain SL, Simpson MA, Pullabhatla V, Tonda R, et al. An in-depth characterization of the major psoriasis susceptibility locus identifies candidate susceptibility alleles within an HLA-C enhancer element. PLoS One. 2013;8:e71690.
11. Benhadou F, Mintoff D, Del Marmol V. Psoriasis:Keratinocytes or Immune Cells – Which Is the Trigger?Dermatology. 2019;235:91-100.
12. Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. 2014;5:5621.
13. Bonifacio KM, Kunjravia N, Krueger JG, Fuentes-Duculan J. Cutaneous expression of A disintegrin-like and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5) in psoriasis goes beyond melanocytes. J Pigment Disord. 2016;3:244.
14. Arakawa A, Siewert K, Stohr J, Besgen P, Kim SM, Ruhl G, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. 2015;212:2203-12.
15. Fuentes-Duculan J, Bonifacio KM, Hawkes JE, Kunjravia N, Cueto I, Li X, et al. Autoantigens ADAMTSL5 and LL37 are significantly upregulated in active psoriasis and localized with keratinocytes, dendritic cells and other leukocytes. Exp Dermatol. 2017;26:1075-82.
16. Lande R, Gregorio J, Facchinetti V, Chatterjee B, Wang YH, Homey B, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564-9.
17. Conrad C, Meller S, Gilliet M. Plasmacytoid dendritic cells in the skin:to sense or not to sense nucleic acids. Semin Immunol. 2009;21:101-9.
18. Ganguly D, Chamilos G, Lande R, Gregorio J, Meller S, Facchinetti V, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983-94.
19. Groves RW, Rauschmayr T, Nakamura K, Sarkar S, Williams IR, Kupper TS. Inflammatory and hyperproliferative skin disease in mice that express elevated levels of the IL-1 receptor (type I) on epidermal keratinocytes. Evidence that IL-1-inducible secondary cytokines produced by keratinocytes in vivo can cause skin disease. J Clin Invest. 1996;98:336-44.
20. Kim BY, Choi JW, Kim BR, Youn SW. Histopathological findings are associated with the clinical types of psoriasis but not with the corresponding lesional psoriasis severity index. Ann Dermatol. 2015;27:26-31.
21. Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond). 2011;120:99-120.
22. Zailaie MZ. Epidermal hydrogen peroxide is not increased in lesional and non-lesional skin of vitiligo. Arch Dermatol Res. 2017;309:31-42.
23. Khalifa E. Sharquie AAN. Follicular vitiligo:the present clinical status ollicular vitiligo:the present clinical status. Our Dermatology Online. 2016.
24. Cabrera R, Recule F, Hojman L, Larrondo J. Follicular vitiligo:dermatoscopic features of a new subtype of vitiligo. An Bras Dermatol. 2019;94:120-21.
25. Alikhan A, Felsten LM, Daly M, Petronic-Rosic V. Vitiligo:a comprehensive overview Part I. Introduction, epidemiology, quality of life, diagnosis, differential diagnosis, associations, histopathology, etiology, and work-up. J Am Acad Dermatol. 2011;65:473-91.
26. Lee AY, Kim NH, Choi WI, Youm YH. Less keratinocyte-derived factors related to more keratinocyte apoptosis in depigmented than normally pigmented suction-blistered epidermis may cause passive melanocyte death in vitiligo. J Invest Dermatol. 2005;124:976-83.
27. Sharquie KE. The histology and immunopathology of vitiligo. PhD Thesis. 1981.
28. Lee DJ, Modlin RL. Breaking tolerance–another piece added to the vitiligo puzzle. J Invest Dermatol. 2005;124:xiii-xv.
29. Namazi MR. Neurogenic dysregulation, oxidative stress, autoimmunity, and melanocytorrhagy in vitiligo:can they be interconnected?Pigment Cell Res. 2007;20:360-3.
30. Sharquie KE, Mehenna SH, Naji AA, Al-Azzawi H. Inflammatory changes in vitiligo:stage I and II depigmentation. Am J Dermatopathol. 2004;26:108-12.
31. Bakry OA, Shoeib M, El Kady N, Attalla S. Re-appraisal of Keratinocytes’Role in Vitiligo Pathogenesis. Indian J Dermatol. 2018;63:231-40.
32. Lee AY. Role of keratinocytes in the development of vitiligo. Ann Dermatol. 2012;24:115-25.
33. Sviderskaya EV, Wakeling WF, Bennett DC. A cloned, immortal line of murine melanoblasts inducible to differentiate to melanocytes. Development. 1995;121:1547-57.
34. Kitamura R, Tsukamoto K, Harada K, Shimizu A, Shimada S, Kobayashi T, et al. Mechanisms underlying the dysfunction of melanocytes in vitiligo epidermis:role of SCF/KIT protein interactions and the downstream effector, MITF-M. J Pathol. 2004;202:463-75.
35. Swope VB, Abdel-Malek Z, Kassem LM, Nordlund JJ. Interleukins 1 alpha and 6 and tumor necrosis factor-alpha are paracrine inhibitors of human melanocyte proliferation and melanogenesis. J Invest Dermatol. 1991;96:180-5.
36. Lee CS, Park M, Han J, Lee JH, Bae IH, Choi H, et al. Liver X receptor activation inhibits melanogenesis through the acceleration of ERK-mediated MITF degradation. J Invest Dermatol. 2013;133:1063-71.
37. Wagner RY, Luciani F, Cario-Andre M, Rubod A, Petit V, Benzekri L, et al. Altered E-cadherin levels and distribution in melanocytes precede clinical manifestations of vitiligo. J Invest Dermatol. 2015;135:1810-19.
38. Cai Y, Fleming C, Yan J. New insights of T cells in the pathogenesis of psoriasis. Cell Mol Immunol. 2012;9:302-9.
39. Yen H, Chi CC. Association between psoriasis and vitiligo:A systematic review and meta-analysis. Am J Clin Dermatol. 2019;20:31-40.
40. Sharquie KE, Salman HA, Yaseen AK. Psoriasis and vitiligo are close relatives. Clin Cosmet Investig Dermatol. 2017;10:341-45.
41. Park JM, Kim HJ, Bae BG, Park YK. A case of concurrent vitiligo and psoriasis. Ann Dermatol. 2009;21:330-3.
42. Menter A, Boyd AS, Silverman AK. Guttate psoriasis and vitiligo:anatomic cohabitation. J Am Acad Dermatol. 1989;20:698-700.
43. Dhar S, Malakar S, Dhar S. Colocalization of vitiligo and psoriasis in a 9-year-old boy. Pediatr Dermatol. 1998;15:242-3.
44. Papadavid E, Yu RC, Munn S, Chu AC. Strict anatomical coexistence of vitiligo and psoriasis vulgaris–a Koebner phenomenon?Clin Exp Dermatol. 1996;21:138-40.
45. Farber EM, Nickoloff BJ, Recht B, Fraki JE. Stress, symmetry, and psoriasis:possible role of neuropeptides. J Am Acad Dermatol. 1986;14:305-11.
46. Falabella R, Barona MI, Echeverri IC, Alzate A. Substance P may play a part during depigmentation in vitiligo. A pilot study. J Eur Acad Dermatol Venereol. 2003;17:355-6.
47. Victor FC, Gottlieb AB, Menter A. Changing paradigms in dermatology:tumor necrosis factor alpha (TNF-alpha) blockade in psoriasis and psoriatic arthritis. Clin Dermatol. 2003;21:392-7.
48. Morelli JG, Norris DA. Influence of inflammatory mediators and cytokines on human melanocyte function. J Invest Dermatol. 1993;100:191S-95S.
49. Zhang S, Liu S, Yu N, Xiang L. RNA released from necrotic keratinocytes upregulates intercellular adhesion molecule-1 expression in melanocytes. Arch Dermatol Res. 2011;303:771-6.
50. Martinez-Esparza M, Jimenez-Cervantes C, Solano F, Lozano JA, Garcia-Borron JC. Mechanisms of melanogenesis inhibition by tumor necrosis factor-alpha in B16/F10 mouse melanoma cells. Eur J Biochem. 1998;255:139-46.
51. Tobin AM, Kirby B. TNF alpha inhibitors in the treatment of psoriasis and psoriatic arthritis. BioDrugs. 2005;19:47-57.
52. Moellmann G, Klein-Angerer S, Scollay DA, Nordlund JJ, Lerner AB. Extracellular granular material and degeneration of keratinocytes in the normally pigmented epidermis of patients with vitiligo. J Invest Dermatol. 1982;79:321-30.
53. Toosi S, Orlow SJ, Manga P. Vitiligo-inducing phenols activate the unfolded protein response in melanocytes resulting in upregulation of IL6 and IL8. J Invest Dermatol. 2012;132:2601-9.
54. Zhu KJ, Lv YM, Yin XY, Wang ZX, Sun LD, He SM, et al. Psoriasis regression analysis of MHC loci identifies shared genetic variants with vitiligo. PLoS One. 2011;6:e23089.
55. Jadotte YT, Janniger CK. Pityriasis alba revisited:perspectives on an enigmatic disorder of childhood. Cutis. 2011;87:66-72.
56. Kim D, Lockey R. Dermatology for the allergist. World Allergy Organ J. 2010;3:202-15.
57. Berth-Jones J, Eczema, Lichenification, Prurigo and Erythroderma, in Rook’s Textbook of Dermatology. 2010. p. 1-51.
58. Miazek N, Michalek I, Pawlowska-Kisiel M, Olszewska M, Rudnicka L. Pityriasis alba–common disease, enigmatic entity:Up-to-date review of the literature. Pediatr Dermatol. 2015;32:786-91.
59. Blessmann Weber M, Sponchiado de Avila LG, Albaneze R, Magalhaes de Oliveira OL, Sudhaus BD, Cestari TF. Pityriasis alba:a study of pathogenic factors. J Eur Acad Dermatol Venereol. 2002;16:463-8.
60. In SI, Yi SW, Kang HY, Lee ES, Sohn S, Kim YC. Clinical and histopathological characteristics of pityriasis alba. Clin Exp Dermatol. 2009;34:591-7.
61. Vinod S, Singh G, Dash K, Grover S. Clinico epidemiological study of pityriasis alba. Indian J Dermatol Venereol Leprol. 2002;68:338-40.
62. Macmillan A, Rook A. Vitiligo with a raised rim in atopic subjects. Br J Dermatol. 1971;85:491.
63. Pinto FJ, Bolognia JL. Disorders of hypopigmentation in children. Pediatr Clin North Am. 1991;38:991-1017.
64. Khalifa E. Sharquie AAN, Haitham M. Salmo. Pityriasis alba versus vitiligo. Journal of the Saudi Society of Dermatology &Dermatologic Surgery. 2013;17:51-54.
65. Wolf R, Wolf D, Trau H. Pityriasis alba in a psoriatic location. Acta Derm Venereol. 1992;72:360.
66. Stankler L, Ewen SW. Follicular psoriasis. Br J Dermatol. 1981;104:153-6.
67. Sharquie KE, Noaimi AA, Mijthab ZM. Chronic Scalp Folliculitis versus Acne Vulgaris (Observational Case Series Study). Journal of Clinical &Experimental Dermatology Research. 2012;3.
68. Thomas M, Khopkar US. Keratosis pilaris revisited:is it more than just a follicular keratosis?Int J Trichology. 2012;4:255-8.
69. Sharquie KE, Noaimi AA, Hameed AF. Lichen planopilaris is a common scarring alopecia among Iraqi population. Journal of Cosmetics, Dermatological Sciences and Applications. 2013:35-39.
70. Thomas LJ, Dadzie OE, Francis N, Morar N. Follicular psoriasis – A forgotten entity?The Open Dermatology Journal. 2010:95-96.
71. Groscurth P. Anatomy of sweat glands. Curr Probl Dermatol. 2002;30:1-9.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-0265-2040 http://orcid.org/0000-0002-0265-2040 |

Comments are closed.