Coexistence of Parry–Romberg syndrome and lichen sclerosus et atrophicus in an adolescent female: A rare combination
Theodora Douvali1, Georgios Emmanouil 1, Maria Gerochristou1, Maria Gerodimou1, Eleftheria Tampouratzi2, Vasiliki Chasapi1
1, Maria Gerochristou1, Maria Gerodimou1, Eleftheria Tampouratzi2, Vasiliki Chasapi1
1Andreas Syggros Hospital of Cutaneous & Venereal Diseases, Athens, Greece,2Tzaneio Hospital of Piraeus, Piraeus, Greece
Corresponding author: Georgios Emmanouil, MD
How to cite this article: Douvali T, Emmanouil G, Gerochristou M, Gerodimou M, Tampouratzi E, Chasapi V. Coexistence of Parry Romberg syndrome and lichen sclerosus et atrophicus in an adolescent female: A rare combination. Our Dermatol Online. 2021;12(3):297-300.
Submission: 26.12.2020; Acceptance: 16.05.2021
DOI:10.7241/ourd.20213.16
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Parry–Romberg syndrome (PRS) is a rare form of localized scleroderma mainly affecting children and young adults, characterized by progressive hemifacial atrophy due to shrinkage and degeneration of tissues beneath the skin. Lichen sclerosus et atrophicus (LSA) is a rare chronic inflammatory skin disease that mainly affects preadolescent and perimenopausal females with anogenital and extragenital localization. Herein, we present a case of the coexistence of these two rare entities in a young adolescent female in the lower half of the face. To our knowledge, although there are numerous cases in the literature describing the coexistence of LSA and localized scleroderma, similar cases of the coexistence of PRS and LSA in the same site have not been discussed.
Key words: Parry–Romberg syndrome; lichen sclerosus et atrophicus; extragenital lichen sclerosus; progressive hemifacial atrophy; localized scleroderma
INTRODUCTION
Parry–Romberg Syndrome (PRS) is considered an unusual variant of localized scleroderma [1], characterized by progressive hemifacial atrophy due to shrinkage and degeneration of tissues beneath the skin [2–5]. Cerebral disturbance of fat metabolism has been proposed as a primary cause. The syndrome usually manifests itself in the first decades of life, with a higher prevalence in females than males [4]. PRS may lead to serious deformities and, for this reason, immediate intervention is required in order to stop the progression of the disease.
Lichen sclerosus et atrophicus (LSA) is a rare chronic inflammatory skin disease that mainly affects preadolescent and perimenopausal females with anogenital and extragenital localization [6]. These patients develop characteristic shiny and pale atrophic plaques with concomitant pruritus and a burning sensation. LSA may coexist with other autoimmune diseases [7].
To the best of our knowledge, the cooccurrence of extragenital LSA with PRS in the same lesion is being discussed here for the first time in the literature, although the autoimmune nature of these two diseases may justify the simultaneous appearance of these disorders in the same patient.
CASE REPORT
A 14-year-old female presented with a five-month history of a solitary, sharply demarcated, pale, atrophic patch in the middle of the jaw, in conjunction with asymmetry and downward deviation of the lower lip (Fig. 1) and tongue (Fig. 2). A dermatoscopic examination of the lesion revealed findings compatible with those of follicular plugging. During the follow-up, within a four-month time period, there was a new, gradual appearance of pale plaques submandibularly and in the lower-left middle of the face (Fig. 3), while the patient also mentioned anogenital pruritus with vaginal discharge, erythema, and the partial disappearance of the clitoris. According to the patient’s history, she suffered from dyslipidemia under medication. A recent immunization against HPV viruses had also been conducted.
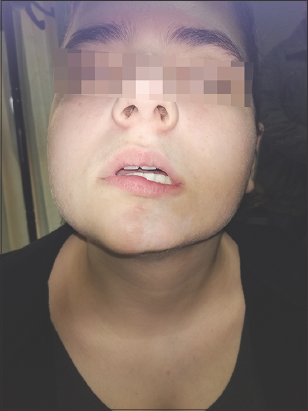 |
Figure 1: A solitary, sharply demarcated, pale, atrophic patch in the middle of the jaw in conjunction with asymmetry and downward deviation of the lower lip. |
 |
Figure 2: Deviation of the patient’s tongue toward the side of the lesion. |
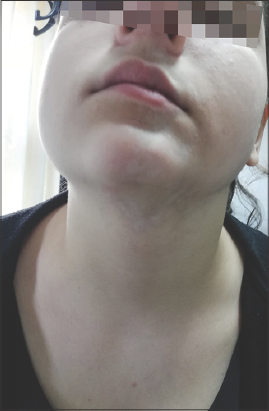 |
Figure 3: Pale plaques affecting the submandibular region and the lower-left middle of the patient’s face. |
Due to the progressive deterioration and tooth tearing reported, a punch biopsy from the plaque in the jawline was performed. The biopsy revealed results compatible with localized scleroderma, revealing diffuse dermal fibrosis with atrophy of the exocrine glands and moderate periadnexal lymphocytic infiltration (Fig. 4). A supplementary MRI brain scan was performed, without any pathological brain findings, apart from high signal intensity in cube flair images in the left condylar process of the mandible. Vaginal smear cultivation and IgG and IgM serum antibodies against Borrelia burgdorferi were negative. A wide serum immunological profile was held, including nRNP/Sm, Sm, SS-A, Ro-52, SS-B, Scl-70, PM-Scl-100, Jo-1, CENP B, PCNA, dsDNA, nucleosomes, histones, rib P-prot, and AMA M2, and gave normal results. The patient’s clinical features regarding the white skin atrophy of the jawline in conjunction with the histopathological results and the partial loss of the clitoris led to the diagnosis of the nosological entities of the co-existence of PRS and LSA.
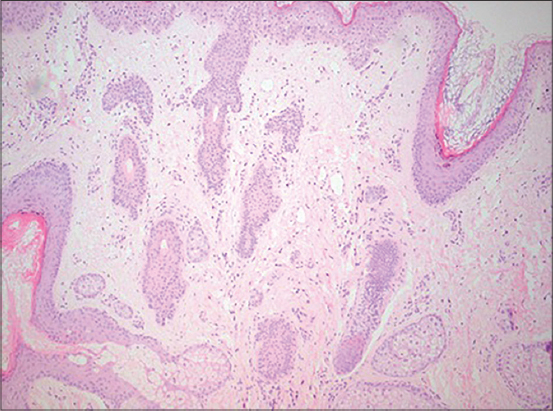 |
Figure 4: Diffuse dermal fibrosis with atrophy of the exocrine glands and moderate periadnexal lymphocytic infiltration. |
Firstly, topical corticosteroids and topical calcineurin inhibitors were administrated. However, there was a gradual worsening of the anogenital symptoms and the face lesions. Thus, a supplementary administration of prednisolone 20 mg per day for four weeks per os in combination with methotrexate 12.5 mg once a week per os, in conjunction with folic acid 5 mg a day before and a day after the initiation of methotrexate. During the first two-week follow-up, liver dysfunction was revealed, making it impossible to increase the dose of the per os regimens. After a period of four weeks, signs of partial disease inactivity were observed and the dose of the corticosteroid was tapered. Our patient, during 1.5 years of follow-up, has stabilized without deterioration of the skin lesions with a dose of methotrexate 12.5 mg weekly in combination with topical agents.
DISCUSSION
PRS is a rare, typically unilateral, acquired disease affecting mainly the face, leading to the atrophy of the skin, the subcutaneous layer, the musculature, and, in some cases, the cartilage and the bony structures of the facial zone. In some cases, there is also the involvement of the arms, trunk, and legs [4].
The age of onset seems to be the first twenty years of life, although a late onset has also been described. PRS progresses slowly and is self-limited, typically waning after 2–10 years and becoming stationary. PRS has a higher prevalence in females[4].
Atrophy of the skin and the underlying tissues is mainly unilateral and inferior to the forehead, usually involving the dermatomes of the branches of the fifth cranial nerve [2]. The structures of the oral cavity may also be affected.
Neurological and ocular manifestations and maxillofacial complications are the most common extracutaneous findings [3–5].
The aim of the treatment of PRS is to halt the disease activity. The most common drug administered is methotrexate, with doses varying from 0.3 to 1 mg/kg/week for 1–2 years, in order to avoid a relapse. Because of the delayed effect of methotrexate, high doses of systematic corticosteroids (prednisone 1 mg/kg/day) are also employed for three months, with a tapering dose at the third month [1,4].
A variant of scleroderma known as en coup de sabre (ECDS) shares common clinical and histopathological findings with PRS. Some authors attempted to separate the two entities by mainly clinical criteria, for instance, by the lack of previous induration, inflammation, and cutaneous atrophy in patients with PRS in contrast with the preceded hyperpigmentation and induration of the skin in ECDS that affects mainly the area above the brow [8]. Nevertheless, even in the same patient, overlapping of the clinical features of these two diseases may be observed [4,8]. A significant overlap of the histopathological features of ECDS and PRS is also present.
Connective tissue fibrosis, adnexal atrophy, and white mononuclear cell infiltrates are present not only in ECDS, but also in some of PRS biopsies [4].
Numerous authors suggest that these two entities fall into the same spectrum of the disease.
In our case, there was the overlap and coexistence of PRS not with a type of scleroderma, such as ECDS, but with LSA, which is a separate autoimmune disease.
LSA is a chronic inflammatory autoimmune disease, commonly affecting the anogenital region. Extragenital LSA may also be found. It affects mostly females, with a bimodal age distribution. The average age of diagnosis is 7.6 years in girls and 60 years in women [4].
The common clinical findings are ivory-white, shiny patches with concomitant atrophy, appearing predominantly in the anogenital area with an hourglass-like appearance. Pruritus or a burning sensation are often present. Morbidity may also occur in vulvar LSA [3]. Although there is often the coexistence of LSA and localized scleroderma (LS) in many patients, the relationship between the two disorders remains controversial. In some cases, the two disorders may appear in the same lesion [7].
Most of the time, histopathology may distinguish the two entities. In LSA, the epidermis is hyperkeratotic with hydropic degeneration of the basal layer. The elastic fibers are characteristically decreased [7].
Both LSA and PRS are of an unknown etiology and pathogenesis.
The possible role of HPV virus association in the development of LSA has been studied [9]. Our patient had undergone a recent immunization against HPV viruses.
Both diseases have a high rate of associated autoimmune diseases [3,10], and trauma seems to play a role in some cases [8].
CONCLUSION
To the best of our knowledge, this is the first case of the coexistence of PRS and LSA in the same extragenital site. Although the literature describes numerous cases of the concomitance of LSA with LS, similar cases of the coexistence of PRS and LSA in the same site have not been discussed. The present findings support the autoimmune origin of the two diseases, the close relation of LS with PRS, and the theory that both entities lie in the same spectrum of a disease.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. El-Kehdy J, Abbas O, Rubeiz N. A review of Parry-Romberg syndrome. J Am Acad Dermatol. 2012;67:769-84.
2. Singh D, Singla C, Malhotra SK, Kaur A. Parry-Romberg syndrome in an elderly male:A rare case report. Our Dermatol Online. 2017;8:333-5.
3. Pope E, Laxer RM. Diagnosis and management of mrphea and lichen sclerosus and atrophicus in children. Pediatr Clin North Am. 2014;61:309-19.
4. Tolkachjov SN, Patel NG, Tollefson MM. Progressive hemifacial atrophy:A review. Orphanet J Rare Dis. 2015;10:39.
5. Jampani K, Nagaraja A, Peddireddy S, Kumar VS. Linear scleroderma:A series of all clinical variants. Our Dermatol Online. 2014;5:185-7.
6. Parajuli N, Poudel P, Tiwari S, Karki A. Linear lichen sclerosus of the face. Our Dermatol Online. 2020;11:379-81.
7. Lis-Święty A, Mierzwińska K, Wodok-Wieczorek K, Widuchowska M, Brzezińska-Wcisło L. Co-existence of lichen sclerosus and localized scleroderma in female monozygotic twins. J Pediatr Adolesc Gynecol. 2014;27:e133-6.
8. Lehman TJ. The Parry Romberg syndrome of progressive facial hemiatrophy and linear scleroderma en coup de sabre. Mistaken diagnosis or overlapping conditions?J Rheumatol. 1992;19:844-5.
9. Nasca MR, Lacarrubba F, Micali G. Human papillomavirus infection and lichen sclerosus:Coincidence or link?Int J Dermatol. 2018;57:617-8.
10. Kreuter A, Kryvosheyeva Y, Terras S, Moritz R, Möllenhoff K, Altmeyer P, et al. Association of autoimmune diseases with lichen sclerosus in 532 male and female patients. Acta Derm Venereol. 2013;93:238-41.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-5415-9514 http://orcid.org/0000-0002-5415-9514 |

Comments are closed.