Destombes and Rosai-Dorfman disease of the anterior abdominal wall – Extra nodal sinus histiocytosis with massive lymphadenopathy
Hubert Daisley Jr 1, Sailaja Golamari1, Lilly Paul2, Deandra Thomas-Romain3, Dawn Meyers1, Martina Daisley4
1, Sailaja Golamari1, Lilly Paul2, Deandra Thomas-Romain3, Dawn Meyers1, Martina Daisley4
1Department of Pathology, General Hospital San Fernando, Trinidad, West Indies, 2Department of Pathology, Scarborough General Hospital, Tobago, West Indies, 3Department of Surgery, Scarborough General Hospital, Tobago. West Indies, 4Department of Accident and Emergency, Scarborough General Hospital. Tobago. West Indies
Corresponding author: Prof. Hubert Daisley Jr, MB BS DM FRCPE MFFLM
Submission: 20.09.2020; Acceptance: 10.12.2020
DOI: 10.7241/ourd.20211.19
Cite this article: Daisley Jr H, Golamari S, Paul L, Thomas-Romain D, Meyers D, Daisley M. Destombes and Rosai-Dorfman disease of the anterior abdominal wall – Extra nodal sinus histiocytosis with massive lymphadenopathy. Our Dermatol Online. 2021;12(1):72-75.
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Destombes–Rosai–Dorfman disease—often simply referred to, in the literature, as Rosai–Dorfman disease (RDD)—is a rare, nonmalignant disorder of histiocyte proliferation typically involving the cervical lymph nodes. A subset of patients with RDD, however, display extranodal manifestations highly variable in presentation, more challenging to diagnose, and less likely to spontaneously regress when compared to the nodal manifestations. This study describes the case of a young African male presenting himself with multiple nodules involving the anterior abdominal wall, who was found to have extranodal RDD. The current mode of diagnosis and the clinical management of RDD are reviewed.
Key words: Destombes–Rosai–Dorfman disease; Extranodal; Histiocytic proliferation
INTRODUCTION
Sinus histiocytosis with massive cervical lymphadenopathy is a non-neoplastic proliferative disease of histiocytes of unknown etiology. It is commonly referred to as Rosai–Dorfman disease (RDD), although it was Destombes who first reported it [1]. The nodal presentation of RDD is fairly common and poses little difficulty in diagnosis. However, the extranodal presentation is uncommon and often presents a diagnostic challenge to pathologists. We report a case and present the modern tools available to aid in such diagnosis.
CASE REPORT
A 29-year-old African male presented himself to the Accident and Emergency department with a two-year history of an initial pustule on the left upper abdomen. During that period, however, the pustule resolved and was replaced by multiple, small, nonfluid-filled, erythematous papules in a hyperpigmented area. The lesions were nonpruritic. There were no constitutional symptoms.
An examination revealed a 3 × 2.5 cm area of hyperpigmented skin on the left upper abdomen with multiple nontender, firm papules. There was no lymphadenopathy.
Laboratory investigations performed showed a complete blood count with normal parameters. Liver and renal function tests were essentially normal, except for hypoglobulinemia of 2.74 g/dL (range: 3.2–3.9 g/dL) (Table 1).
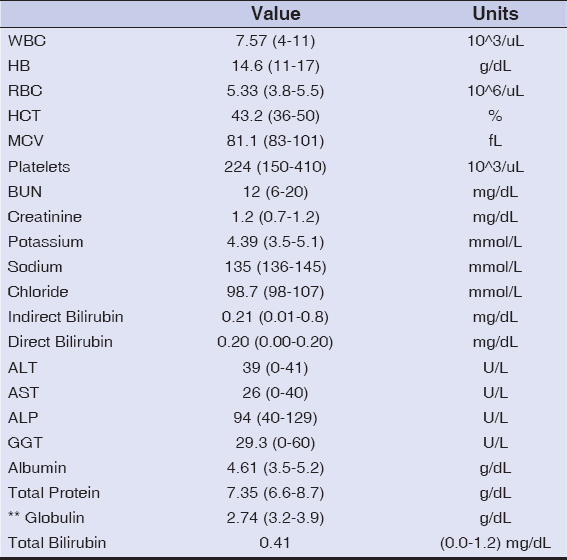 |
Table 1: The hematological and biochemical profi le of the patient |
An excision biopsy of the lesion was done and sent for histological examination.
Histology
Received for histology was a 3.5 × 3 × 2.4 cm ellipse of skin with multiple firm tan papules. The cut sections revealed firm, rubbery, faint-pink tissue (Fig. 1).
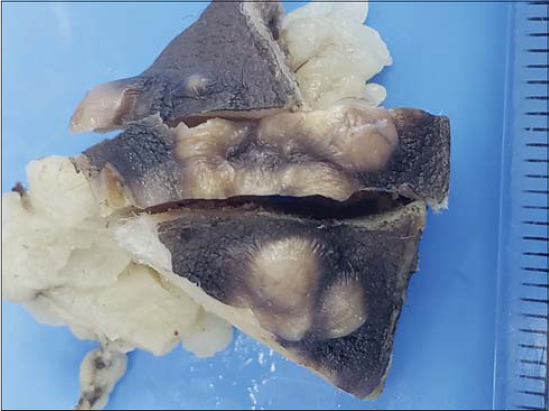 |
Figure 1: The excised lesion from the abdominal wall. |
The histopathology of the skin lesion mimicked those seen in lymph nodes with sinus histiocytosis with massive lymphadenopathy.
The lesion was composed mainly of a well-circumscribed lymphohistiocytic infiltrate involving the dermis and subcutaneous tissue (Fig. 2a). The lesional histiocytes constituted a large central hyperchromatic nucleus set in abundant, pale, water-clear cytoplasm and single distinct nucleoli. Within the lesional histiocytes, emperipolesis was evident, which constituted intact lymphocytes and neutrophils in the cytoplasm (Figs. 2b and 2c). Mature lymphocytes and numerous plasma cells and neutrophils were also present between histiocytes. Mitosis was rare and necrosis was absent. The overlying epidermis showed hyperkeratosis, parakeratosis, and atrophy of rete ridges. Several binucleated histiocytes resembling Reed–Sternberg (RS) cells were seen, but there was a complete lack of expression of the markers CD15 and CD30, which made Hodgkin lymphoma quite unlikely.
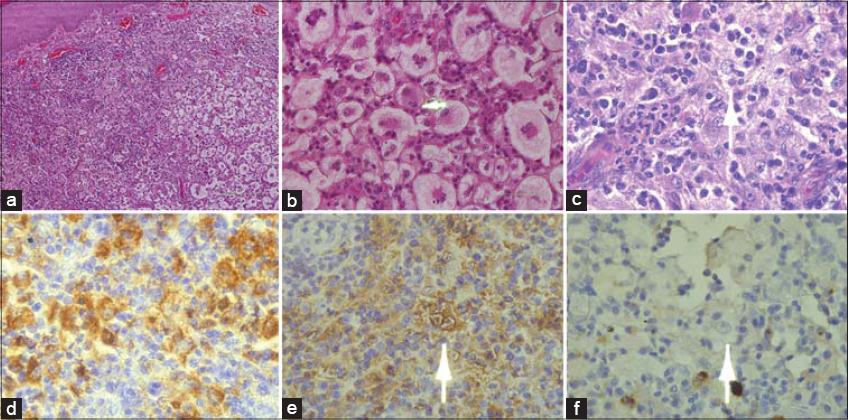 |
Figure 2: (a) The lymphohistiocytic infiltrate in the dermis. (b) Emperipolesis of lymphocytes and neutrophils in a histiocyte. (c) Emperipolesis (H&E, 40×). (d) CD68 positivity (40×). (e) An S100-positive binucleated histiocyte of Destombes–Rosai–Dorfman disease. (f) A CD15-negative histiocyte demonstrating that the histiocyte is not a lacunar cell of Hodgkin lymphoma. |
Immunohistochemical stains confirmed the diagnosis of extranodal RDD by showing positivity for CD68 and S100 (Figs. 2d and 2e).
A few binucleated histiocytes resembled those of Reed-Sternberg cells (RS cells) were seen, however there was a complete lack of expression of the markers CD15 (Fig. 2f) and CD30, which makes Hodgkin Lymphoma quite unlikely.
The overall histological picture of the abdominal lesion was that described by Rosai and Dorfman in lymph nodes with sinus histiocytosis with massive lymphadenopathy.
DISCUSSION
The disease entity first reported in French by Destombes in 1965 [1] was further characterized by Rosai and Dorfman in 1969 [2]. Hence, it would be fitting for the disease to bear the name of the original researcher. Destombes–Rosai–Dorfman disease is seen in all ages and ethnicities, but with a slight predominance in young males.
The classic presentation includes fever and painless massive cervical lymphadenopathy, the latter showing sinus histiocytosis with emperipolesis as the hallmark of the disease [3]. Other lymph node groups may be involved.
RDD is a benign proliferative histiocytic disorder of unknown etiology, self-limiting in most instances. The extranodal manifestations may present themselves in any site, with cases reported in the bone, eyelid, skin, central nervous system, lungs, and other sites with or without nodal involvement [4–8].
The extranodal lesions often present a challenge to histopathologists because of the broad differentials for lymphohistiocytosis, and all these histiocytic disorders share the common pathological findings of abnormal proliferation of histiocytes.
Disorders warranting a differential diagnosis with RDD include lipogranulomatosis, Langerhans cell histiocytosis, Hodgkin disease, metastatic carcinoma, malignant melanoma, and Erdheim–Chester disease.
A lack of Touton giant cells eliminates the diagnosis of lipogranuloma. Langerhans cell histiocytosis, similarly to RDD, demonstrates lymphohistiocytic proliferation and positive expression of S100 and CD1a. However, cells of Langerhans cell histiocytosis lack lymphophagocytosis and, ultrastructurally, reveal the presence of the characteristic rod-shaped Birbeck granules. Lacunar cells of nodular sclerosis, a form of Hodgkin disease, may on occasion be confused with RDD cells. However, Reed–Sternberg cells and their variants do not exhibit emperipolesis. They are S100-protein-negative, CD15-positive, and CD30-positive. Negative staining for HMB-45 and pankeratin differentiate RDD from melanoma and metastatic carcinoma [9].
Erdheim–Chester disease is a rare non-Langerhans cell histiocytosis and a true histiocytic neoplasm characterized by multisystemic proliferation of mature histiocytes in a background of inflammatory cells [10]. Cutaneous infiltrates in Erdheim–Chester disease contain xanthogranulomata with large Touton giant cells, which are not reactive cells, but part of the neoplastic process. In addition, there is the common background of inflammatory cells of foamy histiocytes, lymphocytes, and neutrophils, as observed in RDD. Emperipolesis is not a feature of Erdheim–Chester disease.
The clinical and pathological features in our case fit those described by Destombes, Rosai, and Dorfman, with emperipolesis being the hallmark and the distinguishing feature [3].
Surgical excision of these lesions in extranodal sites is the treatment of choice, although there have been reports of recurrence after excision [11]. Therapy with acitretin, thalidomide, methotrexate, and steroids has shown promising results in cases that recurred after surgical excision [12,13]. Complete resolution of the disease has been reported with siltuximab therapy [14]. Further research needs to be done to better understand the underlying pathophysiology of RDD. In doing so, clinical management would become less problematic.
CONCLUSION
The extranodal manifestations of Destombes–Rosai–Dorfman disease (RDD) present numerous diagnostic and therapeutic challenges, as it may occur in any organ or site. However, the use of immunotyping aids the pathologist in making the diagnosis. There is much to learn about the etiology of RDD for effective therapy to be possible.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Destombes P. [Adenitis with lipid excess, in children or young adults, seen in the Antilles and in Mali. (4 cases)]. Bull Soc Pathol Exot Filiales. 1965;58:1169-75.
2. Rosai J, Dorfman RF:Sinus histiocytosis with massive lymphadenopathy:A newly recognized benign clinicopathological entity. Arch Pathol. 1969;87:63-70.
3. Rastogi V, Sharma R, Misra SR, Yadav L, Sharma V. Emperipolesis – A review. J Clin Diagn Res. 2014;8:ZM01-2.
4. Komaragiri M, Sparber LS, Santos-Zabala ML, Dardik M, Chamberlain RS. Extranodal Rosai-Dorfman disease:A rare soft tissue neoplasm masquerading as a sarcoma. World J Surg Oncol. 2013;11:63.
5. Sandoval-Sus JD, Sandoval-Leon AC, Chapman JR, Velazquez-Vega J, Borja MJ, Rosenberg S, Lossos A, Lossos IS. Rosai-Dorfman disease of the central nervous system:Report of 6 cases and review of the literature. Medicine (Baltimore). 2014;93:165-75.
6. Quentin M, Samina B, Philippe M, Emile J et al. Lung involvement in Destombes-Rosai-Dorfman disease:Clinical and radiological features and response to the MEK inhibitor cobimetinib. Chest. 2019;157:323-33.
7. Leal PA, Adriano AL, Breckenfeld MP, Costa IS, de Sousa AR, Gonçalves Hde S. Rosai-Dorfman disease presenting with extensive cutaneous manifestation – Case report. An Bras Dermatol. 2013;88:256-9.
8. Singh S, Kumar A, Pandey S, Kumar R, Singh I, Kumari N. Isolated Langerhans cell histiocytosis masquerading as intradural extramedullary meningioma:Review on histiocytic disorders of spine. J Pediatr Neurosci. 2019;14:46-51.
9. Kim JY, Shim HK, Kim MR. A case of xxtranodal Rosai-Dorfman disease presenting as an isolated mass on the base of the tongue in a 57-year-old woman. Am J Case Rep. 2020;21:e925716.
10. Ozkaya N, Rosenblum MK, Durham BH, Pichardo JD, Abdel-Wahab O, Hameed MR, et al. The histopathology of Erdheim-Chester disease:A comprehensive review of a molecularly characterized cohort. Mod Pathol. 2018;31:581-97.
11. Lin CK, Tsai YD. Nonresectable thoracic Rosai-Dorfman Disease:A case report and review of the literature. World Neurosurg. 2019;132:309-13.
12. Nadal M, Kervarrec T, Machet MC, Petrella T, Machet L. Cutaneous Rosai-Dorfman disease located on the breast:Rapid effectiveness of methotrexate after failure of topical corticosteroids, acitretin and thalidomide. Acta Derm Venereol. 2015;95:758-9.
13. Shahidi-Dadras M, Hamedani B, Niknezhad N, Ghilizadeh N. Rosai-Dorfman disease successfully treated with thalidomide:A case report. Dermatol Ther. 2019;32:e13005.
14. Lee H, King G, Garg K, Pan Z, Tobin J, Robinson WA. Successful treatment of disseminated Rosai-Dorfman disease with siltuximab. Haematologica. 2018;103:e325-8.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-7337-8050 http://orcid.org/0000-0002-1785-4315 http://orcid.org/0000-0002-9061-9565 http://orcid.org/0000-0001-7708-0000 http://orcid.org/0000-0001-9675-6512 http://orcid.org/0000-0001-9785-3648 http://orcid.org/0000-0002-7337-8050 http://orcid.org/0000-0002-1785-4315 http://orcid.org/0000-0002-9061-9565 http://orcid.org/0000-0001-7708-0000 http://orcid.org/0000-0001-9675-6512 http://orcid.org/0000-0001-9785-3648 |

Comments are closed.