Giant idiopathic pyoderma gangrenosum at an unusual site with highly elevated c-ANCA levels: A rare association
Lovleen Kaur , Mohita Mahajan, Parul Chojer, Bharat Bhushan Mahajan, Suresh Kumar Malhotra
, Mohita Mahajan, Parul Chojer, Bharat Bhushan Mahajan, Suresh Kumar Malhotra
Department of Dermatology, Venereology and Leprosy, Government Medical College, Amritsar, India
Corresponding author: Dr. Lovleen Kaur
Submission: 13.05.2020; Acceptance: 01.08.2020
DOI: 10.7241/ourd.20211.15
Cite this article: Kaur L, Mahajan M, Chojer P, Mahajan BB, Malhotra SK. Giant idiopathic pyoderma gangrenosum at an unusual site with highly elevated c-ANCA levels: A rare association. Our Dermatol Online. 2021;12(1):58-61.
Citation tools:

Copyright information
© Our Dermatology Online 2021. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Pyoderma gangrenosum (PG) is a rare neutrophilic disorder with an incidence rate of 3–10 cases per million per year, characterized by classically painful and aseptic ulcers, which may be associated with underlying systemic diseases. The pathergy reaction is seen in one-fourth of patients with PG. Accurate and timely diagnosis is crucial, as PG is known for its rapid progression. The management of PG is challenging and depends on its severity and rate of progression. An underlying systemic involvement should be sought even in spite of no symptoms. Herein, we report a case of giant pyoderma gangrenosum involving almost the entire left buttock with exceptionally raised c-ANCA levels, but no underlying systemic abnormality. The patient reported intense pain, rapid progression of the ulcers, an inability to perform daily activities, was significantly morbid and pathergy-positive. Aggressive and early management is required in cases such as this. A dramatic response was achieved with a combination of cyclosporine, dapsone, and methylprednisolone pulses.
Key words: Pyoderma gangrenosum; Neutrophilic dermatoses; ANCA; Pathergy
INTRODUCTION
Pyoderma gangrenosum (PG) is a rare neutrophilic dermatosis characterized by noninfectious ulcers, usually involving the lower extremities, with an estimated incidence rate of 3–10 cases per million per year [1]. PG is associated with underlying systemic diseases in around 50% of cases [2]. Modified immunological response and altered neutrophilic chemotaxis have been proposed as the underlying mechanism of pathogenesis. Pathergy is seen in one-fourth of cases of PG [3]. Herein, we report a case of giant pyoderma gangrenosum on an atypical site—the left buttock—with highly elevated c-ANCA levels, treated with multidrug therapy.
CASE REPORT
A 28-year-old female presented herself to our dermatology outpatient department with a history of recurrent progressive ulcerations involving the lower extremities and left buttock. The ulcerations had shown two years prior as a single nodular abscess over the right ankle, which had been incised and drained by a surgeon. Pathergy developed at the incision site to form a painful nonhealing ulcer. As the ulcer expanded, the patient noted simultaneous development of new ulcers bilaterally on the shins, ankles, and the dorsum of both feet within several days. While some of the ulcers healed with scarring, others progressively enlarged despite continuous treatment with oral steroids, doxycycline, and azathioprine elsewhere. Intermittent remissions and relapses of the lesions occurred during these two years.
Later, the patient developed three small ulcers on the left buttock while on treatment with azathioprine, oral steroids, and minocycline for two consecutive months. The edge of one ulcer was biopsied, after which the ulcers enlarged and coalesced to form a large necrotic ulcer within a period of three days, thus, again, demonstrating the presence of a pathergy reaction. No further medical or surgical history of significance was recorded.
An examination revealed a giant oval ulcer 23 × 17 cm in size covered with a necrotic brown crust on the left buttock (Figs. 1a and 1b). Manipulation produced purulent exudate and oozing of the blood. Another deep-seated ulcer, 2 × 3 cm in size and adjacent to the initial, was present medially. Its base was not fixed to the underlying structures. Multiple discrete erythematous-to-violaceous warm and tender nodules—several with central suppuration—were present over both lower limbs, predominantly over the thighs, indicating primary lesions. A single left inguinal lymph node 2 × 2 cm in size was present. Multiple healed atrophic cribriform scars were evident on both the shins and the dorsum of the feet (Figs. 2a and 2b). A systemic examination was unremarkable.
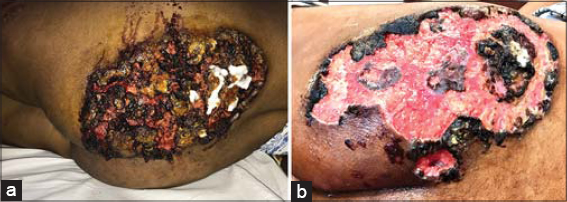 |
Figure 1: (a) The initial presentation as a giant necrotic crusted lesion covering almost the entire left buttock (day 1). (b) The ulcer covered with frank purulent exudate after desloughing (day 3). |
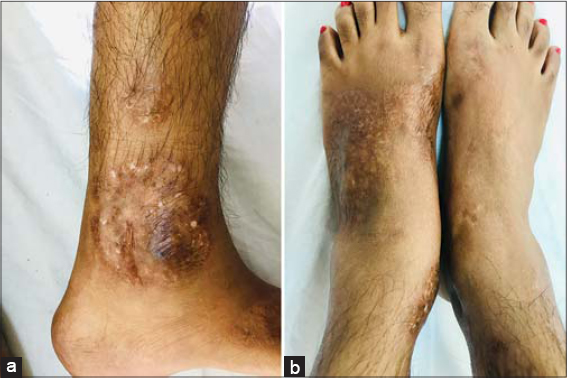 |
Figure 2: (a) Atrophic cribriform-like scarring on the lateral aspect of the right foot and the right lower leg. (b) The dorsum of the left foot and the medial aspect of the left ankle. |
Routine and specific investigations revealed hemoglobin at 6.6 g/dL, a total leukocyte count of 13,000 with neutrophilia (90%), ESR at 102, C-reactive protein at 49.4, and rheumatoid factor at 298.7. c-ANCA (antineutrophil cytoplasmic antibody) levels were as high as 76.0 units/mL. ANA (antinuclear antibody), anti-CCP (anti-cyclic citrullinated peptide), p-ASCA (anti-saccharomyces cerevisiae antibody), and p-ANCA (perinuclear-staining ANCA) were normal. Fecal occult blood was absent. Colonoscopy was inconclusive of inflammatory bowel disease (IBD), showing several tiny ileal ulcerations with the rest of the intestinal mucosa completely normal. A pus culture revealed growth of Escherichia coli. Tubercular and fungal cultures revealed no growth. A deep skin biopsy from the edge of the ulcer revealed partially treated vasculitis, endothelial swelling, intravascular thrombi, and large neutrophilic aggregates in the subcutaneous tissue, consistent with the diagnosis of PG (Figs. 3a and 3b). No grains or AFB were seen.
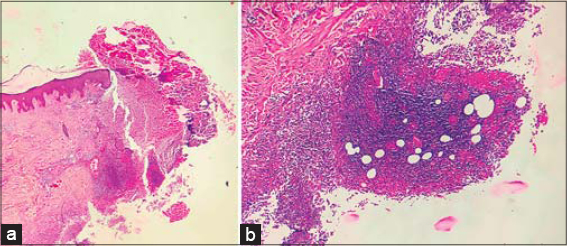 |
Figure 3: (a) A photomicrograph of the edge of the ulcer (gross view) (H&E, scanner view). (b) Intravascular thrombi and large neutrophilic aggregates in the subcutaneous tissue (H&E, 10×). |
The patient was given a blood transfusion and IV antibiotics. Once the secondary infection was controlled, methylprednisolone pulses for five days, oral cyclosporine 200 mg/day, and dapsone 100 mg/day were initiated. Anticipating the risk of pathergy, surgical debridement was discouraged. Desloughing with hydrogen peroxide and daily sterile dressings were performed. Within two weeks of initiating treatment, the purulent exudate reduced markedly and the ulcer began re-epithelizing with healthy granulation tissue at the floor (Fig. 4). Complete healing was observed in two months, although the atrophic hyperpigmented scar was still present (Fig. 5). The patient was, however, fully satisfied. Drugs were gradually tapered and stopped after six months. Currently, the patient is off cyclosporine and dapsone, and has had no recurrence after nine months of continuous follow-ups. The levels of ESR, CRP, and c-ANCA (4.0 units/mL) have returned to normal.
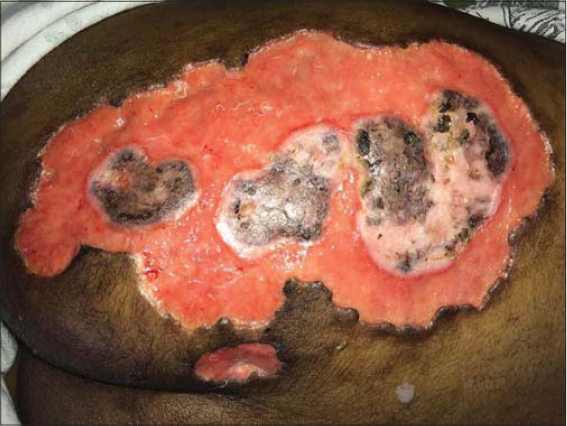 |
Figure 4: A response two weeks after therapy, with epithelization evident in the center and healthy granulation tissue at the floor of the ulcer. |
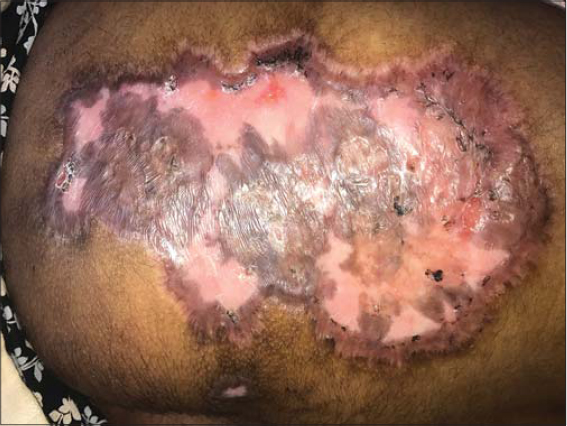 |
Figure 5: The nearly complete resolution of the ulcer after two months of therapy. |
DISCUSSION
Pyoderma gangrenosum ulcers are classically painful and aseptic, although superinfection may eventually occur, probably, as in our case, because of an odd anatomical site. This ulcer, on removal of the necrotic crust, showed a purulent base with undermined asymmetrical borders. Extension of the lesion on surgical incision was observed twice, indicating a pathergy reaction. Multiple old healed cribriform scars again favored the diagnosis of PG. Systemic associations of ulcerative colitis, Crohn’s disease, rheumatoid arthritis, hematological malignancies, and monoclonal gammopathy have been reported [2].
Skin manifestations in both classical and localized Wegener’s granulomatosis (WG) have been reported in around 50% of cases [4]. They may present themselves as papulonecrotic, urticarial, vesicular lesions, palpable purpura or necrotizing ulcers resembling pyoderma gangrenosum [5]. It must be noted that initial presentations in the form of PG-like lesions have been reported in 10% of cases of WG [4]. Although c-ANCA levels were severely raised, no manifestation suggestive of upper and lower airway and renal involvement were found in this case to fulfil the classical triad of Wegener’s granulomatosis (WG).
Other than the former, ANCA-associated vasculitis (AAV) has been reported with neutrophilic dermatoses (ND). Indeed, several clinical conditions, such as inflammatory bowel diseases (IBDs), can exhibit both ND and ANCA, especially their anti-MPO subtypes [6]. However, the specific targets of these antibodies, as of now, have not been identified. One study suggests that, in cases of neutrophilic dermatosis, the patient may be tested for ANCA and, if positive, careful attention should be paid to observe signs suggestive of systemic vasculitis [7]. Another study, conducted by Kawakami et al., proves the pathologic role of ANCA beyond vasculitis and suggests that c-ANCA may be directly or indirectly pathogenic in PG and/or may be produced because of neutrophilic activation and apoptosis in cases with PG, resulting in a self-amplifying loop [8]. Our patient displayed highly elevated c-ANCA levels, but no sign of WG or IBD was discovered.
Therapy in cases with PG is a matter of debate, as there is no uniformly effective treatment. The aim is to treat the underlying cause. In a rapidly progressive disease with significant morbidity, an aggressive treatment approach to halt the immunological process is crucial. Hence, methylprednisolone pulses, followed by cyclosporine, for its quick onset of action, and dapsone, for its antineutrophilic and steroid-sparing effects are preferred in rapidly progressing cases such as ours, in which such a regimen dramatically improved the outcome and expedited recovery in two months with sustained remission. However, regular follow-ups are necessary to detect signs of recurrence; otherwise, rare cases may display this as an early manifestation of Wegener’s granulomatosis or ulcerative colitis.
CONCLUSION
Pyoderma gangrenosum, a type of neutrophilic dermatosis, oftentimes causes significant agony to the patient. PG is typically noninfectious, painful, and rapidly progressive, and so demands aggressive management with immunosuppressants, as in our case. Surgical maneuvers should be discouraged because of the high risk of pathergy. Common sites are the lower limbs, but PG may present itself at any site, making diagnosis problematic. A thorough systemic examination and a battery of investigations should be performed in search for an underlying systemic abnormality. c-ANCA, although considered highly specific to Wegener’s granulomatosis, may also be a possible marker for ulcerative colitis or AAV associated with PG; hence, long-term follow-ups are obligatory because pyoderma gangrenosum may be an initial presentation of either of these underlying disorders.
Consent
The examination of the patient was conducted according to the principles of the Declaration of Helsinki.
The authors certify that they have obtained all appropriate patient consent forms, in which the patients gave their consent for images and other clinical information to be included in the journal. The patients understand that their names and initials will not be published and due effort will be made to conceal their identity, but that anonymity cannot be guaranteed.
REFERENCES
1. Cozzani E, Gasparini G, Parodi A. Pyoderma gangrenosum:a systematic review. G Ital Dermatol Venereol. 2014;149:587-600.
2. Rodríguez-Zúñiga MJM, Heath MS, Gontijo JRV, Ortega-Loayza AG. Pyoderma gangrenosum:a review with special emphasis on Latin America literature. An Bras Dermatol. 2019;94:729-43.
3. Ahronowitz I, Harp J, Shinkai K. Etiology and management of pyoderma gangrenosum:a comprehensive review. Am J Clin Dermatol. 2012;13:191-211.
4. Nasir N, Ali SA, Mehmood Riaz HM. Cutaneous ulcers as initial presentation of localized granulomatosis with polyangiitis:a case report and review of the literature. Case Rep Rheumatol. 2015;2015:517025.
5. Chhabra S, Minz RW, Rani L, Sharma N, Sakhuja V, Sharma A. Immune deposits in cutaneous lesions of Wegener’s granulomatosis:predictor of an active disease. Indian J Dermatol. 2011;56:758-62.
6. Elkadri AA, Stempak JM, Walters TD, Lal S, Griffiths AM, Steinhart AH, et al. Serum antibodies associated with complex inflammatory bowel disease. Inflammat Bowel Dis. 2013;19:1499-505.
7. de Boysson H, Silva NM, de Moreuil C, Néel A, de Menthon M, Meyer O, et al. Neutrophilic dermatoses in antineutrophil cytoplasmic antibody-associated vasculitis:a French multicenter study of 17 cases and literature review. Medicine. 2016;95:e2957.
8. Kawakami T, Kawanabe T, Saito C, Kannari M, Mizoguchi M, Nagafuchi H, et al. Clinical and histopathologic features of 8 patients with microscopic polyangiitis including two with a slowly progressive clinical course. J Am Acad Dermatol. 2007;57:840-8.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0003-3862-5228 http://orcid.org/0000-0003-3862-5228 |

Comments are closed.