An XPC and XPA genetic study on xeroderma pigmentosum patients in a Moroccan population
Hanane Bay Bay 1, Asmae Rasso1, Niema Aqil1, Leila Bouguenouch1, Karim Ouldim2, Sara Elloudi1, Zakia Douhi1, Fatima Zahra Mernissi1
1, Asmae Rasso1, Niema Aqil1, Leila Bouguenouch1, Karim Ouldim2, Sara Elloudi1, Zakia Douhi1, Fatima Zahra Mernissi1
1Department of Dermatology, CHU Hassan II Fez, Morocco, 2Department of Genetics, Hassan II University Hospital Fez, Morocco
Corresponding author: Prof. Hanane Bay Bay, MD PhD
Submission: 16.02.2020; Acceptance: 28.04.2020
DOI: 10.7241/ourd.20204.1
Cite this article: Bay Bay H, Rasso A, Aqil N, Bouguenouch L, Ouldim K, Elloudi S, Douhi Z, Mernissi FZ. An XPC and XPA genetic study on xeroderma pigmentosum patients in a Moroccan population. Our Dermatol Online. 2020;11(4):335-339.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Background: Xeroderma pigmentosum (XP) is a rare hereditary disease characterized by hypersensitivity to UV radiation due to alterations in the nucleotide excision repair (NER) pathway. The XPA and XPC gene mutations are most common in North African countries. Our goal was to perform a molecular study on patients with XP followed in our northeastern Moroccan region to determine their genetic profile.
Materials and methods: We explored the nonsense (c.682C> T, p.Arg228X) mutation at the XPA gene and a two-base pair deletion (c.1643_1644delTG or p.Val548Ala fsX25) at the XPC gene level with the molecular PCR sequencing technique. Subsequently, the relationship between the mutations found and the symptomatic and progressive features was analyzed.
Results: In the course of our work, the alterations of the two XPA and XPC genes responsible for xeroderma pigmentosum in a sample of 24 index cases belonging to 22 unrelated families were characterized, revealing 14 cases of XPC and 6 cases of XPA. The study on the correlation between genotypes and phenotypes in our study showed that neurological involvement was significant in XPA patients and that these XPA patients develop malignant skin tumors earlier than XPC patients.
Conclusions: In our XP population, the alterations of the XPA and XPC genes responsible for xeroderma pigmentosum in a sample of 24 index cases belonging to 22 unrelated families were characterized, revealing 14 cases of XPC and 6 cases of XPA. Neurological involvement was significant in XPA patients and these XPA patients were found to develop malignant skin tumors earlier than XPC patients.
Key words: Xeroderma pigmentosum, XPA gene, XPC gene, Morocco
INTRODUCTION
Xeroderma pigmentosum (XP) is a rare genetic disease with autosomal recessive inheritance first described in 1870 by Hungarian dermatologist Moritz Kaposi, who coined the term xeroderma, Latin for “dry or parched skin.” The term pigmentosum underlines the pigmentary disturbances in patients suffering from this disease [1,2]. Its autosomal recessive transmission explains its relative frequency in countries where consanguinity is high and the size of families is considerable, for instance, in Morocco [3]. In 1968, Cleaver demonstrated a deficiency of UV repair of deoxyribonucleic acid (DNA) in XP cells [4], which produces hypersensitivity to ultraviolet rays and, consequently, a high risk of developing signs of “Heloderma” and poikiloderma associated with xerosis and skin fragility, sometimes evolving to infected and trailing ulcerations, as well as cutaneous malignant tumors and oculars at an early age [5]. Indeed, it has been shown that the clinical heterogeneity of this disease is linked to the existence of alteration in the genes belonging to the various classical complementation groups—XP-A to XP-G—which are distinguished by certain symptomatic and evolutionary peculiarities. The product of each of these genes plays a specific role in the nucleotide excision resynthesis (NER) DNA repair pathway. The human XPA and XPC gene mutations are most common in Maghreb countries [3,6,7].
The Goal of the Study
In this work, we performed a molecular study of the XPA and XPC genes on XP patients followed in our region to determine their genetic profile. Subsequently, the relationship between the mutations found and the symptomatic and progressive features was analyzed.
MATERIALS AND METHODS
This was a cross-sectional, prospective, descriptive, and analytical study conducted from June 2017 through January 2018 at the Dermatology Department of Hassan II University Hospital in Fez, which drains patients from the northeastern region of Morocco.
All patients followed for xeroderma pigmentosum (XP) were included in the collaboration with the Genetics Department. We described the epidemiological and clinical features of these patients, then explored the two most common mutations in North African countries with the molecular PCR sequencing technique. We, first, explored the XPC gene mutation at exon 9 (c.1643_1644delTG or p.Val548Ala fsX25) in all patients, followed by the XPA gene at exon 6 (c. 682C> T, p.Arg228X) for those in whom the first XPC mutation was negative.
These XP patients were examined in a day hospital. Sampling was performed in two 5-mL EDTA (ethylenediaminetetraacetic acid) tubes, each of blood, which, then, were sent to the Genetics Department for DNA extraction and PCR sequencing. The tubes were stored at -20°C for later use. The results of molecular sequencing were obtained after two weeks on average.
After obtaining the sequence of exon 9 of the XPC gene and that of exon 6 of the XPA gene, a bioinformatic analysis of the sequences was performed—in particular, identification of the similarities between the query sequence and the sequence from the database by the Blast software. Subsequently, the relationship between the mutations found and the symptomatic and progressive features of the patients was analyzed.
Ethics Statement
Ethics approval was obtained from the ethics committees of the University Hospital Center Hassan II in Fez, Morocco. All patients, or their parents, were informed of the conditions related to the study and gave their informed consent for the study and for publication.
RESULTS
We collected a sample of 24 patients from 22 unrelated families, two of whom had two children with AIDS. The average age was 15 years with extremes ranging from 2 to 63 years. The majority of patients (n = 20) were under 25 years of age. The patients studied had a sex ratio (M/F) of 0.6, with 9 (37.5%) males and 15 females (62.5%). The patients came from different parts of Morocco, mainly from the northwest, with a predominance of the Fez region (n = 14). The majority of the patients were of rural origin (n = 16). 62.5% (n = 15) of the patients had first-degree consanguinity, 16.7% (n = 4) had second-degree consanguinity, while 20.8% (n = 5) had no consanguinity.
All our patients had a dark phototype, with 79.2% (n = 19) phototype IV, 16.7% (n = 4) phototype III, and 4.2% (n = 1) phototype V. The mean age of onset of early symptoms of the disease was 42 months, with extremes ranging from one month up to 20 years. Photophobia and cutaneous photosensitivity were consistent signs in all our XP patients. Dermatological examinations found a poikiloderma appearance in all patients. The majority of the patients had benign skin tumors (n = 19); these tumors were made of nevi (Fig. 1), warts, seborrheic keratoses, pyogenic granulomas, keratoacanthomas, and ruby angiomas. The patients had precancerous pigmented and unpigmented actinic keratose lesions (Fig. 2), as well as various malignant skin tumors, such as basal cell carcinomas (BCC), squamous cell carcinomas (EC) (Fig. 3), and melanomas. No cases of sarcoma or lymphoma were noted. Ocular involvement of allergic or bacterial conjunctivitis was noted in 8 patients (33%).
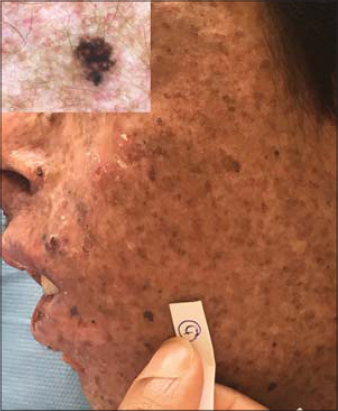 |
Figure 1: Junctional naevus in a patient with XP. |
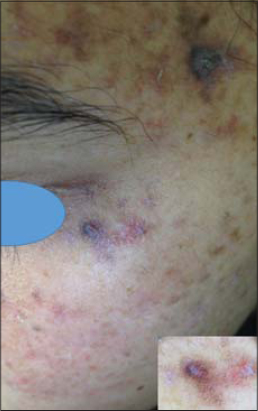 |
Figure 2: Actinic keratosis in a patient with XP. |
 |
Figure 3: Squamous cell carcinoma in a patient with XP. |
Three patients developed ocular tumors of squamous cell carcinoma, responsible, in two of them, for bilateral blindness and, in one patient, for unilateral enucleation. Two patients had failure to thrive; one had rickets; and one had high blood pressure. A molecular study of the XPC gene revealed that the mutation was found in 14 patients with common deletion of the T and G nucleotides affecting exon 9 of the XPC gene (c.1643_1644delTG or p.Val548Ala fsX25), 12 in the homozygous state and 2 in the heterozygous state.
A molecular study on XP brothers from two families revealed the presence of deletion of exon 9 of the XPC gene.
As for the analysis of the XPA gene performed on the 10 patients in whom the XPC gene analysis did not reveal pathogenic abnormalities, six patients had a nonsense mutation (c.682C> T, p.Arg228X) serving at the level of the XPA gene and 4 patients had no abnormality (c.682C> T, p.Arg228X). The results indicated that neurological involvement was significant in XPA patients. (P = 0.023). The same genetic group showed earlier malignant skin tumors (of less than 6 years) with the occurrence of acute reaction after exposure to the sun, when compared to patients with XPC (P = 0.000).
DISCUSSION
Xeroderma pigmentosum (XP) is a rare autosomal recessive genetic disorder that affects the Moroccan population with a relatively high incidence rate when compared with Europe. We were able to characterize 24 XP patients—the largest group analyzed in Morocco to date. The sex ratio of our patients was 0.6, with a predominance of women; this is consistent with the results of a 2010 study by Soufir et al. [3]. Inbreeding was found in 79.2% of cases. The average age of onset was 42 months in the Soufir study, as in ours [3]. The diagnosis of the disease was reached later, at an average age of 9.5 years.
Clinical manifestations were photophobia, photosensitivity, pruritus, poikiloderma, and xerosis in all patients, in agreement with the results reported in the literature [3,4]. These XP patients had benign cutaneous tumors: nevi, warts, seborrheic keratoses, botriomyomas, keratoacanthomas, and ruby angiomas, with variable frequency. Malignant skin tumors appeared in 79% of the XP patients, with CBC in 70.8% of cases, EC in 37.5% of cases, and melanoma in 12.5% of cases. This high frequency of cutaneous tumors might be explained by the delay of the diagnosis of the disease and, therefore, the delay in initiating photoprotective measures. Soufir et al., in 2010, and Senhaji et al., in 2012, observed the same results [3,6]. In our study, the average age of development of the first skin cancer was 9.3 years; all patients who had developed skin cancer developed at least one cancer other than a melanoma (CBC and/or CE) in areas exposed to the sun. In 2000, Chavanne et al. reported that the average age of development of the first skin cancer was 11.7 years in 7 XPC patients from southern Europe [8]. In 2012, Schafer et al. reported a mean premature age of 7.1 years in 16 XPC patients from Germany [9]. The low frequency of neurological involvement in our series was similar to that reported in the Soufir study [3].
In the literature, ocular involvement in XP is variable and depends on the complementation group; this includes photophobia, conjunctivitis, keratitis, entropion, ectropion, and ocular tumors.
The XPC and XPA groups have a higher risk of developing this type of damage than the XPE group. In our study, the results matched those found in the literature without any difference in the severity of ocular involvement, whatever the genetic group [10]. PCR sequencing revealed that the nonsense mutation (c.682C> T, p.Arg228X) at the XPA gene level was present in 25% (n = 6) of our XP patients. Common deletion of the nucleotides T and G affecting exon 9 of the XPC gene (c.1643_1644delTG or p.Val548Ala fsX25) was found in 58% (n = 14) of the patients, 12 in the homozygous state and 2 in the heterozygous state. In these two heterozygous patients, another mutation ought to be sought to consider compound heterozygosity.
A molecular study by Soufir et al. of 66 unrelated families in the Maghreb region showed that 85% of the patients had XPC gene mutations; among them, 87% shared the founding mutation (approx. 1643 1644delTG). 12% of the XP patients had mutations in the XPA gene, with a mutation frequency (c.682C> T) of approximately 87.5% [3]. In an Algerian series, the XPA mutation (c.682C> T) was present in 2 of 19 patients (10.5%) and the XPC mutation (1643 1644delTG) was present in 17 of 19 patients (89.5%) in the homozygous state [11]. In a study conducted in Casablanca, the XPC mutation (c 1643 1644delTG) was estimated to account for more than 76% of cases of XP in Moroccan patients [12]. Another study conducted at the same institution involving XP patients with neurological involvement revealed the presence of the nonsense mutation (c.682C> T) in the homozygous state at the level of the XPA gene in 78 patients [3].
The frequency of XPA mutation was previously described in North Africa, but it is more common in Japan [3]. The common homozygous XPC mutation (c.1643_1644delTG) has been described in North Africa, mainly in patients from Tunisia, and three patients from Italy, Egypt, and Africa, respectively [7,8]. Two compound heterozygous patients (XP132BE and XP30BE) were from the United States and Honduras. In our study, there was no significant difference between XPA and XPC patients in the age of onset of the disease. In contrast to the literature describing an earlier age of onset of XPA [10,13].
The prevalence of malignant skin tumors is higher in XPC patients than in XPA patients, which might be explained by the early onset of XPA, leading to early photoprotection, thus preventing the occurrence of skin tumors [14,15]. This difference was absent in our study, which may have been due to the delay in the consultation of parents because of the lack of knowledge on this pathology.
CONCLUSION
In our work, the alterations of the XPA and XPC genes responsible for xeroderma pigmentosum in a sample of 24 index cases belonging to 22 unrelated families were characterized, revealing 14 cases of XPC and 6 cases of XPA. Our study on the correlation between genotypes and phenotypes revealed that neurological involvement was significant in XPA patients and that these XPA patients developed malignant skin tumors earlier than XPC patients, which requires earlier implementation of photoprotective strategies to prevent such damage.
Acknowledgments
We are indebted to all patients who participated in this study and gave their consent. We thank all volunteer investigators and the medical staff of the Department of Dermatology and Genetics.
Statement of Human and Animal Rights
All the procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the 2008 revision of the Declaration of Helsinki of 1975.
Statement of Informed Consent
Informed consent for participation in this study was obtained from all patients.
REFERENCES
1. Salissou L, Doulla M, Harouna MZ, Salha I, Timmi N, Sono Abdou AD, Moumouni H, Sani R, Nouhou H. [Xeroderma pigmentosum:Squamous cell carcinoma infi ltrating and disfi guring facial, in a girl of 3 years and a half]. Our Dermatol Online. 2017;8(Suppl. 1):28-31.
2. Crocker HR. Three cases of xeroderma pigmentosum (Kaposi) or atrophoderma pigmentosum. Med Chir Trans. 1884;67:169–188.
3. Soufir N, Ged C, Bourillon A, Austerlitz F, Chemin C, Stary A, et al. A prevalent mutation with founder effect in xeroderma pigmentosum group C from north Africa. J Invest Dermatol. 2010;130:1537–42.
4. Cleaver JE. Defective repair replication of DNA in xeroderma pigmentosum. DNA Repair. 2004;3:1837.
5. Yadalla HKK, Juwariya S. Xeroderma pigmentosum:A bane in developing country – Brief report. Our Dermatol Online. 2014;5:395-7.
6. Senhaji MA, Abidi O, Nadifi S, Benchikhi H, Khadir K, Ben Rekaya M, et al. c.1643_1644delTGXPC mutation is more frequent in Moroccan patients with xeroderma pigmentosum. Arch Dermatol Res. 2013;305:53-7.
7. Diatta BA, Diallo S, Ndiaye M, TouréS, Gaye GW, Dieng MT. A squamous cell carcinoma of the lower lip with a discoid lupus erythematosus. Our Dermatol Online. 2019;10:376-8.
8. Chavanne F, Broughton BC, Pietra D, Nardo T, Browitt A, Lehmann AR, et al. Mutations in the XPC gene in families with xeroderma pigmentosum and consequences at the cell,protein, and transcript levels. Cancer Res. 2000;60:1974–82.
9. Schäfer A, Hofmann L, Gratchev A, Laspe P, Schubert S, Schürer A, et al. Molecular genetic analysis of 16 XP-C patients from Germany:environmental factors predominately contribute to phenotype variations. Exp Dermatol. 2013;22:24-9.
10. Fassihi H, Sethi M, Fawcett H, Wing J, Chandler N, Mohammed S, et al. Deep phenotyping of 89 xeroderma pigmentosum patients reveals unexpectedheterogeneity dependent on the precise molecular defect. Proc Natl Acad Sci U S A. 2016;113:E1236-45.
11. Bensenouci S, Louhibi L, De verneuil H, Mahmoudi K, Saidi-Mehtar N. Diagnosis of Xeroderma Pigmentosum groups A and C by detection of two prevalent mutations in West Algerian population:A rapid genotyping tool for the frequent XPC mutation c.1643_1644delTG. Biomed Res Int. 2016;2016:2180946.
12. Kindil Z, Senhaji MA, Bakhchane A, Charoute H, Chihab S, Nadifi S, et al. Genetic investigation of XPA gene:high frequency of the c.682C>T mutation in Moroccan XP patients with moderate clinical profile. BMC Res Notes. 2017;10:704.
13. Sonnappa UK, Samayam A. Case report on xeroderma pigmentosum with squamous cell carcinoma in a ten year old child. Our Dermatol Online. 2018;9:160-3.
14. Al-Kamel MA. Keratoacanthoma of the nose coexisted with xeroderma pigmentosum in a Yemeni child:A rare case. Our Dermatol Online. 2016;7:419-21.
15. Kgokolo M, Morice-Picard F, Rezvani HR, Austerlitz F, Cartault F, Sarasin A, et al. Xeroderma pigmentosum in South Africa:Evidence for a prevalent founder effect. Br J Dermatol. 2019;181:1070-2.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/000-0003-3455-3810 http://orcid.org/000-0003-3455-3810 |

Comments are closed.