Cutaneous epithelioid angiosarcoma of the head and neck. A case report of an unusual aggressive entity with limited treatment options
Sandra Gonzalez1, Raul Gerardo Mendez 1, Bertha Lissette Sotelo1, Francisco Solano2
1, Bertha Lissette Sotelo1, Francisco Solano2
1Universidad De Guadalajara, Dermatology Service of Civil Hospital of Guadalajara “Fray Antonio Alcalde”, Guadalajara, Mexico. 2Universidad De Guadalajara, Private Practice Pathology, Guadalajara, Mexico
Corresponding author: Dr. Raul Gerardo Mendez Flores, E-mail: drraulmendezf@outlook.com
Submission: 20.01.2020; Acceptance: 18.02.2020
DOI: 10.7241/ourd.20202.12
Cite this article: Gonzalez S, Mendez RG, Sotelo BL, Solano F. Cutaneous epithelioid angiosarcoma of the head and neck. A case report of an unusual aggressive entity with limited treatment options. Our Dermatol Online. 2020;11(2):161-164.
Citation tools:

Copyright information
© Our Dermatology Online 2020. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Cutaneous angiosarcoma (CA) is an uncommon malignant mesenchymal neoplasm with aggressive behavior and a high mortality rate. A rare histological variant of CA is the epithelioid variant angiosarcoma (EA), which is associated with a worse clinical outcome. We report a 74-year-old female with an 8-month history of an ulcerated, poorly defined, painless tumoral mass on her scalp, face, and neck. Her past medical history included rheumatoid arthritis. A panel of immunohistochemical markers positive for vascular markers CD31 and CD34, confirmed the diagnosis. She had an overall 9-month survival since the diagnosis was made. Comorbid diseases and tumoral size make most of CA patients ineligible surgical candidates. Limited treatment options are available and due to its accelerated progression, achieving local control and prevention of metastasis in EA is challenging. We emphasize the need for early diagnosis and a multidisciplinary approach in order to improve survival in these patients.
Key words: Angiosarcoma; Epithelioid; Aggressive; Head; Neck
INTRODUCTION
Cutaneous angiosarcoma (CA) is an uncommon malignant mesenchymal neoplasm with aggressive behavior and high mortality rate. CA is especially rare and make up less than 1% of all sarcomas [1]. VEGF-A is the most studied cytokine and is consistently expressed at higher concentrations in angiosarcomas than in benign vascular or normal-tissue controls [2].
CA usually arises in three characteristic clinical contexts, which include sun-damaged skin of the elderly, at the site of previous radiation treatment, and in lymphedematous limbs (Stewart-Treves syndrome).CA clinical presentation is characterized by large erythematous or violaceous patches, plaques or nodules, commonly affecting the face and scalp [3].
A rare histological variant of CA is the epithelioid variant angiosarcoma (EA). This neoplasm subtype arises mainly in deep soft tissues, however, cases with purely and primarily cutaneous involvement have been reported [4]. Like other angiosarcomas, EA presents as erythematous or violaceous lesions. They are usually ill-defined, infiltrative hemorrhagic tumors. Concerning CA prognosis, tumor necrosis and epithelioid cytomorphology are associated with a worse clinical outcome [5].
Cervical lymph node metastases and widespread dissemination to the visceral organs and the skeleton may develop, and less than 15% of patients survive more than 5 years [6]. Treatment of choice in patients with CA remains surgical resection with or without postoperative radiation therapy [7], however, this is challenging because the tumor often exceeds the clinically apparent margins. In case the tumor is too extensive to consider surgical resection, there is no standard treatment.
CASE REPORT
A 74-year-old Hispanic female presented to our clinic with an 8-month history of a painless tumoral mass on her scalp, face and neck. Initially, she noticed purplish lumps on her neck with increasing nodularity and size and associated erythema with slight desquamation. Extension to her right ear, forehead and bilateral cheeks was noted sequentially. Her past medical history included rheumatoid arthritis for 10 years with poor control.
At examination, multiple, polymorphic, irregular tumoral masses were noted on her face and neck, that converged forming infiltrated plaques with poorly defined edges and ulceration on her right temple, as shown in Fig. 1.
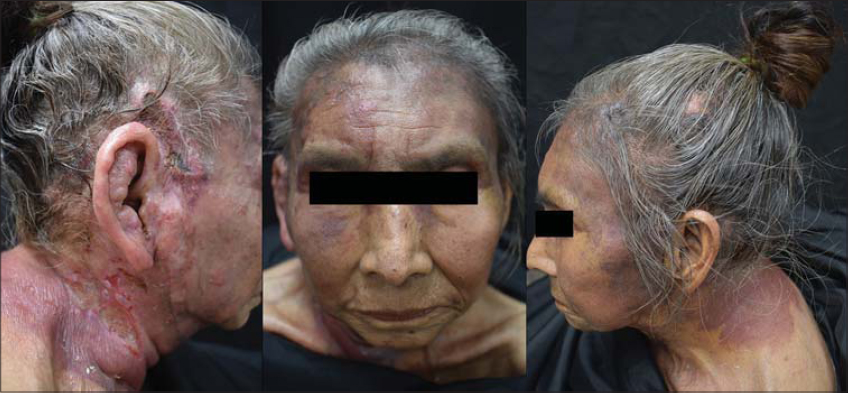 |
Figure 1: Multiple, polymorphic, irregular tumoral masses were noted on her face and neck, that converged forming infiltrated plaques with poorly defined edges and ulceration on her right temple. |
Work up included a CT scan of head and neck that showed a poorly defined mass invading soft tissues in the temporal and parietal area, as well as ipsilateral enlarged cervical lymph nodes.
Two incisional biopsies were analyzed, showing a predominantly vascular neoplasm with interanastomosing vessels lined by epithelioid endothelial cells which dissected collagen bundles, and at the periphery, a large solid sheet of epithelioid cells (Figs. 2 and 3). Tumoral epithelioid cells were polygonal with amphophilic cytoplasm and prominent nuclear atypia.
 |
Figure 2: Tumoral polygonal epithelioid cells with amphophilic cytoplasm and prominent nuclear atypia lining interanastomosing vessels. |
 |
Figure 3: Large solid sheet of epithelioid cells in the periphery of the lesion. |
A panel of immunohistochemical markers was performed that revealed that the neoplastic cells were strongly positive for vascular markers: CD31, CD34, and vimentin (Figs. 4 and 5).
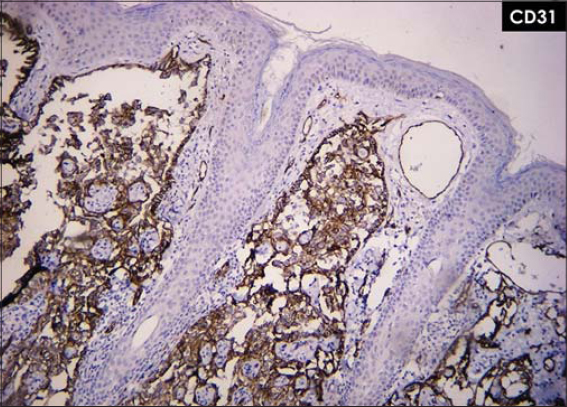 |
Figure 4: Neoplastic cells strongly positive for vascular marker CD31. |
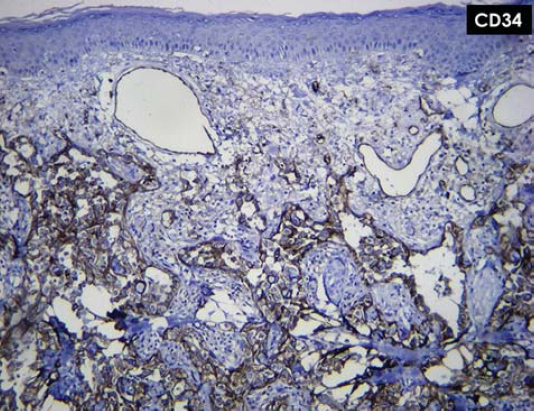 |
Figure 5: Neoplastic cells trongly positive for vascular marker CD34. |
She was diagnosed with a clinical T2 epithelioid angiosarcoma, according to the TNM staging system of the American Joint Committee on Cancer (AJCC).
Due to the extent of the disease and her feeble state, the patient was an ineligible surgical candidate; resection, if performed, would have obliged the removal of almost her entire scalp, from ear to ear. She was referred to the Oncology service which she failed to follow up. Unfortunately, she died 1 month after the diagnosis was made.
DISCUSSION
Age, tumor size, location, follow-up loss and delay in diagnosis prompted our patient fatal outcome. She had an overall 9-month survival since the diagnosis was made.
Most patients present with an important delay in diagnosis. Delays often occur due to clinical variability, with CA appearance mimicking an infectious condition, an angiomatous lesion, or a post-traumatic bruise. Pawlik et al [8] reported that the median time to diagnosis was 5.1 months. The patients who died (58.6%) had an overall median actuarial survival of 28.4 months. Patients who were older than 70 years had a significantly worse median survival (18.2 months).
Tumor (T) classification also was found to be an important prognostic factor that significantly affected overall survival. Clinical T1 disease had a median actuarial survival of 48.7 months, whereas patients with clinical T2 disease had a survival of 11.1 months [8].
A recent meta-analysis with data pooled from 11 studies [9], showed a 5-year survival rate of 33.5%. Their analysis identified age (>70 years), tumor size (>5cm, T2), location (scalp>face) and margin status as predictors of poor prognosis.
Doxorubicin and weekly paclitaxel are both regarded as a preferred option as the first or second line, and these regimens provide a median overall survival of approximately 8 to 12 months [10]. Few reports have described successful treatment of AS using adjuvant chemotherapy with radiotherapy [11].
Treatments with anti-angiogenic antibodies for advanced and aggressive entities like EA had been unsatisfactory. Agulnik et al. [12] reported in a non-randomized phase II trial assessing bevacizumab as monotherapy in angiosarcoma patients; the median progression-free survival (PFS) was 3 months, and the reported best objective response rate was only 8%. Current clinical trials registered at clincaltrials.gov (accessed 15 January 2020) involving CA are evaluating paclitaxel as monotherapy and in combination with radiation therapy, and a relatively new oncolytic virus, talimogene laherparepvec (T-VEC) [13].
Comorbid diseases and tumoral size make most of CA patients’ ineligible surgical candidates. Limited treatment options are available and due to its accelerated progression, achieving local control and prevention of metastasis in EA is challenging. We emphasize the need for early diagnosis and a multidisciplinary approach in order to improve survival in these patients.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
The authors certify that they have obtained all appropriate patient consent forms. In the form the patient(s) has/have given his/her/their consent for his/her/their images and other clinical information to be reported in the journal. The patients understand that their names and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
REFERENCES
1. Rouhani P, Fletcher CD, Devesa SS, Toro JR. Cutaneous soft tissue sarcoma incidence patterns in the U.S.:an analysis of 12,114 cases. Cancer. 2008;113:616-27.
2. Young RJ, Brown NJ, Reed MW, Hughes D, Woll PJ. Angiosarcoma. Lancet Oncol. 2010;11:983-91.
3. Wood A, Mentzel T, van Gorp J, Flucke U, Huschka U, Schneider J, et al. The spectrum of rare morphological variants of cutaneous epithelioid angiosarcoma. Histopathology. 2015;66:856-63.
4. Mobini N. Cutaneous epithelioid angiosarcoma:a neoplasm with potential pitfalls in diagnosis. J Cutan Pathol. 2009;36:362-9.
5. Deyrup AT, McKenney JK, Tighiouart M, Folpe AL, Weiss SW. Sporadic cutaneous angiosarcomas:a proposal for risk stratification based on 69 cases. Am J Surg Pathol. 2008;32:72-7.
6. Abraham JA, Hornicek FJ, Kaufman AM, Harmon DC, Springfield DS, Raskin KA, et al. Treatment and outcome of 82 patients with angiosarcoma. Ann Surg Oncol. 2007;14:1953-67.
7. Ishida Y, Otsuka A, Kabashima K. Cutaneous angiosarcoma:update on biology and latest treatment. Curr Opin Oncol. 2018;30:107-12.
8. Pawlik TM, Paulino AF, McGinn CJ, Baker LH, Cohen DS, Morris JS, et al. Cutaneous angiosarcoma of the scalp:a multidisciplinary approach. Cancer. 2003;98:1716-26.
9. Shin JY, Roh SG, Lee NH, Yang KM. Predisposing factors for poor prognosis of angiosarcoma of the scalp and face:Systematic review and meta-analysis. Head Neck. 2017;39:380-6.
10. Penel N, Italiano A, Ray-Coquard I, Chaigneau L, Delcambre C, Robin YM, et al. Metastatic angiosarcomas:doxorubicin-based regimens, weekly paclitaxel and metastasectomy significantly improve the outcome. Ann Oncol. 2012;23:517–23.
11. De Yao JT, Sun D, Powell AT, Rehmus EH. Scalp angiosarcoma remission with bevacizumab and radiotherapy without surgery:a case report and review of the literature. Sarcoma. 2011;2011:160369.
12. Agulnik M, Yarber JL, Okuno SH, von Mehren M, Jovanovic BD, Brockstein BE, et al. An open-label, multicenter, phase II study of bevacizumab for the treatment of angiosarcoma and epithelioid hemangioendotheliomas. Ann Oncol. 2013;24:257-63.
13. Lalu M, Leung GJ, Dong YY, Montroy J, Butler C, Auer RC, et al. Mapping the preclinical to clinical evidence and development trajectory of the oncolytic virus talimogene laherparepvec (T-VEC):a systematic review. BMJ Open. 2019;9:e029475.
Notes
Source of Support: Nil.
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-5945-6593 http://orcid.org/0000-0002-5945-6593 |

Comments are closed.