Le dermatofibrosarcome de Darier Ferrand (Etude de 49 cas)
[Dermatofibrosarcoma protuberans (Report of 49 cases)]
Fatma Frikha, Madiha Mseddi, Emna Bahloul, Rim Chaabouni, Abderrahmen Masmoudi, Sonia Boudaya, Ichrak Chami, Meriem Amouri, Hamida Turki
Département de Dermatologie Vénérologie, CHU Hédi Chaker, Sfax, Tunisie
Corresponding author: Dr. Fatma Frikha, E-mail: frikhafat@gmail.com
Submission: 24.04.2019; Acceptance: 29.07.2019
DOI:10.7241/ourd.20201.5
Cite this article: Frikha F, Mseddi M, Bahloul E, Chaabouni R, Masmoudi A, Boudaya S, Chami I, Amouri M, Turki H. Le dermatofi brosarcome de Darier Ferrand (Etude de 49 cas) [Dermatofi brosarcoma protuberans (Report of 49 cases)]. Our Dermatol Online. 2020;11(1):25-31
Citation tools:
BibTex | CSV | RIS | Endnote XML
Copyright information
© Our Dermatology Online 2019. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Background: retrospective study to elucidate the epidemio-clinical and pathological characteristics, management and evolutional aspects of DFS with comparison with the data of the literature.
Procedure: a thirty seven-year study of all cases of DFS.
Results: Forty-nine cases of DFS (22 males and 27 females) were diagnosed. Mean age at diagnosis was 48.2 years. The tumor appeared before the age of 15 years in five patients (10%). This was a recurrence in 12% of cases. Five patients reported a history of trauma. The average delay before consultation was 5.6 years. A solitary nodule was the predominant clinical aspect of DFS (59.2%). Mean size was 4.7 cm. The tumor was mainly located on the trunk. The diagnosis of dermatofibrosarcoma protuberans was histologically confirmed in all cases. Immunohistochemical study was achieved in 34 cases and showed positive staining for CD34. The treatment consisted of large surgical excision in all patients. Four patients had revision surgery because of tumor margins. Eight cases (17.4%) presented a local recurrence after a mean delay of 12.5 months.
Conclusion: DFS is a low-malignancy potential skin tumor. Treatment of choice is surgery. The main risk is tumor relapse. A long follow-up is then very important to detect it.
Key words: Darier-Ferrand; Dermatofibrosarcoma protuberans; Treatment; Surgery; Recurrence
RÉSUMÉ
Problématique: étude rétrospective afin de décrire les aspects épidémiologiques, cliniques, anatomopathologiques, thérapeutiques et évolutifs avec comparaison aux données de la littérature.
Matériel et Méthodes: Etude de tous les dossiers des patients ayant présenté un DFS sur une période de 37 ans.
Résultats: Quarante-neuf cas de DFS (22 hommes et 27femmes) avaient un âge moyen de 48,2 ans. La tumeur était apparue avant l’âge de 15 ans chez cinq patients (10%). Il s’agissait d’une récidive dans 12% des cas. La notion d’un traumatisme local était rapportée dans 5 cas. Le délai moyen de consultation était de 5,6 ans. L’aspect clinique prédominant était un nodule unique (59,2%). La taille moyenne était de 4,7 cm. Le tronc était la localisation préférentielle (55,1%). Le diagnostic positif était dans tous les cas confirmé par l’examen anatomopathologique. L’étude immunohistochimique était réalisée dans 34 cas et montrait un marquage des cellules tumorales vis-à-vis du CD34. Le traitement reposait sur l’exérèse chirurgicale. La reprise chirurgicale était indiquée chez quatre malades devant des limites atteintes. Une récidive locale était notée dans huit cas (17,4%) avec un délai moyen de 12,5 mois.
Conclusion: Le DFS est un sarcome de faible degré de malignité. Le traitement chirurgical reste le seul traitement adapté de nos jours. L’évolution du DFS est surtout émaillée de récidives locales. Une surveillance clinique prolongée est nécessaire pour les détecter.
Key words: Darier-Ferrand; Dermatofibrosarcoma protuberans; Treatment; Surgery; Recurrence
INTRODUCTION
Le DFS est une tumeur mésenchymateuse rare, à malignité intermédiaire, caractérisée par son évolution lente, sa forte agressivité locale et des métastases exceptionnelles avec une forte tendance à la récidive. Cette tumeur, dont la fréquence est non négligeable dans les pays africains, pose encore plusieurs problèmes en rapport avec son aspect clinique trompeur évoquant surtout une cicatrice chéloïde (fréquente chez la population africaine) et responsable souvent d’un retard diagnostic. Sa gravité est liée à son agressivité locale et son très haut risque de récidive locale [1–4]. Nous présentons une étude rétrospective de 49 cas de DFS dans le but de décrire ses aspects épidémiologiques, cliniques, anatomopathologiques, thérapeutiques et évolutifs. Nos résultats seront comparés aux données de la littérature.
MATERIELS ET METHODES
Nous avons réalisé une étude rétrospective portant sur 49 cas de DFS histologiquement prouvés (sur biopsie cutanée et/ou sur pièce opératoire), répertoriés au service de dermatologie vénéréologie du centre hospitalier universitaire Hédi Chaker Sfax Tunisie, durant une période de 37 ans (1981-2017). Nous avons étudié les caractéristiques épidémiologiques, cliniques, paracliniques, l’aspect anatomopathologique, la prise en charge thérapeutique et le suivi des patients. Le recueil de données a été réalisé à partir des dossiers cliniques des patients.
RESULTATS
Nos 49 patients étaient répartis en 22 hommes (44,9%) et 27 femmes (55,1%). La moyenne d’âge des patients au moment du diagnostic était de 48,2 ans avec des extrêmes de 17 et 84 ans. L’âge de début de la tumeur variait entre un an et 81 ans avec une moyenne de 39,5 ans. Cinq patients (10%) ont rapporté un début avant l’âge de 15 ans (huit mois, un an, sept ans, 11 ans et 14 ans) et 12 patients un début après l’âge de 50 ans. La notion de traumatisme antérieur était retrouvée dans cinq cas (10,2%). Le délai moyen de consultation était de 5,6 ans avec des extrêmes de trois mois et 30 ans. Six patients (12%) se sont présentés pour une récidive d’une tumeur opérée auparavant, une première récidive dans quatre cas et une deuxième récidive dans deux cas.
La tumeur était nodulaire dans 29 cas (59,2%) (Figs. 1 – 4), multinodulaire dans 16 cas (32,6%) (Figs. 5 et 6), et sous forme d’une plaque infiltrée dans quatre cas (8,2%) (Fig. 7). Elle était ulcérée dans six cas et hémorragique dans trois cas. La taille de la tumeur variait de un cm à 20 cm (moyenne de 4,7 cm). Elle était supérieure à dix cm chez cinq patients (Fig. 5). Le tableau I résume les principales localisations du DFS.
 |
Figure 1: Un petit nodule légèrement pigmenté du dos du poignet. Figure 1: [A small, slightly pigmented nodule on the back of the wrist] |
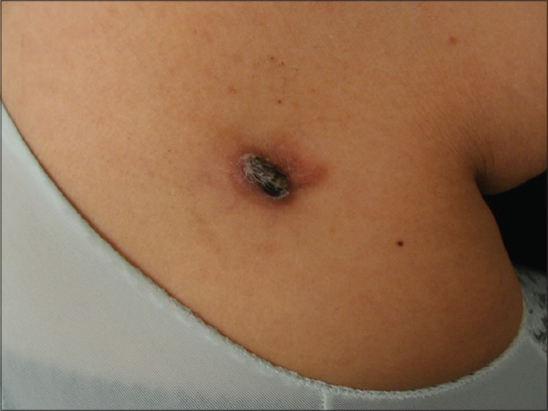 |
Figure 2: Une lésion nodulaire noirâtre infiltrée de la région scapulaire. Figure 2: [A blackish infiltrated nodular lesion in the scapular region] |
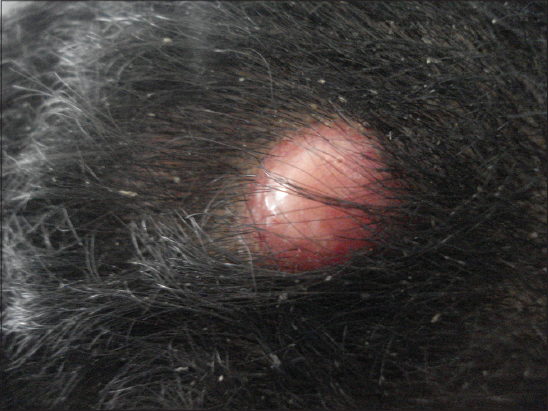 |
Figure 3: Lésion nodulaire érythémateuse du cuir chevelu. Figure 3: [Erythematous nodular lesion of the scalp] |
 |
Figure 4: Lésion nodulaire de la joue. Figure 4: [Nodular lesion of the cheek] |
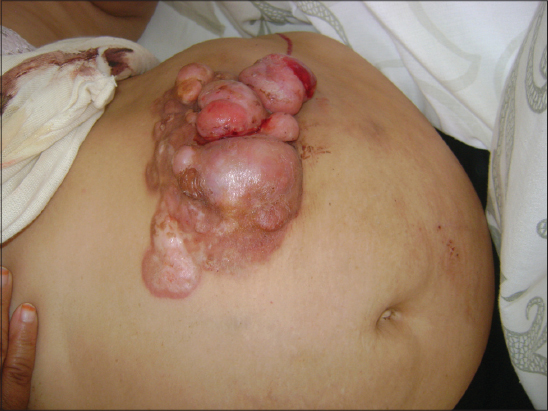 |
Figure 5: Une plaque multi-nodulaire géante du flanc. Figure 5: [A giant multi-nodular plate of the flank] |
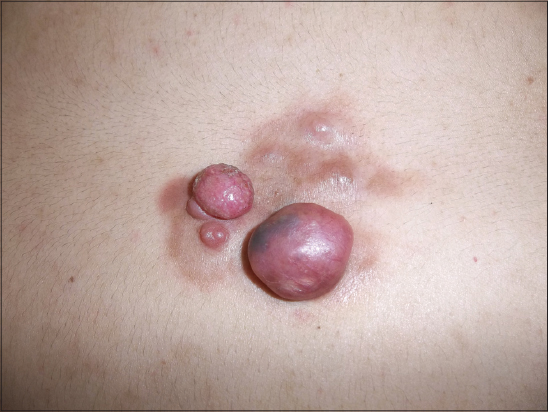 |
Figure 6: Une plaque mutinodulaire de la région lombaire. Figure 6: [A mutinodular plaque of the lumbar region] |
 |
Figure 7: Une plaque infiltrée du dos. Figure 7: [An infiltrated plate of the back] |
 |
Tableau I: Localisation du DFS. Table I: [Localization of Dermatofibrosarcoma protuberans] |
L’examen anatomopathologique a confirmé le diagnostic de DFS dans tous les cas montrant une prolifération dermo-hypodermique de faisceaux courts entrecroisés d’architecture storiforme, composés de cellules fusiformes uniformes peu ou modérément atypiques. L’hypoderme était envahi dans 44 cas (91% des cas) et un envahissement du muscle strié était retrouvé dans deux cas. Deux variantes histologiques particulières étaient retrouvées: DFS myxoïde caractérisé par un stroma myxoïde (deux cas) et DFS pigmenté (tumeur de Bednar) caractérisé par la présence de pigment mélanique dans le cytoplasme des cellules (un cas).
L’étude immunohistochimique, faite pour 34 patients (69%), montrait un marquage diffus (cytoplasmique et membranaire) et intense pour le CD34 dans tous les cas. L’étude cytogénétique n’a pas été réalisée.
Quarante-six patients ont été opérés, l’exérèse chirurgicale était large avec des marges latérales de sécurité de cinq à dix cm chez 12 patients (26%) et de deux à quatre cm chez 34 patients (74%). En profondeur, l’exérèse emportait systématiquement une barrière anatomique saine (l’aponévrose dans 26 cas (56,5%), le périoste dans cinq cas (11%), une parotidectomie exo-faciale était réalisée dans un cas et une excision musculaire dans deux cas). La reprise chirurgicale était indiquée chez quatre malades devant des limites atteintes. La réparation de la perte de substance engendrée par l’exérèse chirurgicale faisait appel aux différents procédés de la chirurgie plastique: une suture directe chez 19 patients (41%), une greffe de peau chez 14 patients (31%) et un lambeau dans 11 cas (24%) et la cicatrisation dirigée chez deux patients (4%). Aucun patient n’a eu une radiothérapie ou une thérapie ciblée. Les récidives locales étaient notées dans huit cas (17,4%) après un délai moyen de 12,5 mois (deux mois à quatre ans). Le délai de récidive était de moins de trois ans dans 87,5% des cas. Ces récidives étaient multiples dans deux cas (un cas avait deux récidives et un cas avait huit récidives).
DISCUSSION
Le DFS, une tumeur à développement intradermique, a été décrite pour la première fois en 1890 par Taylor [5] comme une tumeur sarcomateuse ressemblant à une cicatrice chéloïde. Le DFS fut rapporté ensuite par Darier et Ferrand en 1924, et Hoffman en 1925 [5] qui l’intitule «dermatofibrosarcoma protuberans». En 1951, Pack et Tabah ont publié la première série importante [6] et en 1962, Taylor et Helwig ont établis les caractéristiques histologiques de cette entité [4,5].
L’âge de survenue du DFS se situe généralement entre 20 et 50 ans avec des moyennes oscillant entre 28 et 47 ans selon les auteurs [7–9]. L’âge médian de nos patients était de 39,5 ans (un an à 81 ans). L’affection n’épargne pas les sujets des âges extrêmes. C’est ainsi que sont décrits des cas chez l’enfant, des cas congénitaux et même après 80 ans [9,10]. Les cas pédiatriques (enfant de moins de 15 ans) de DFS sont rares [11–13]. Dans notre série, on avait cinq cas de DFS de l’enfant et 12 cas étaient apparus après l’âge de 50 ans. Aucune relation de cause à effet n’a été démontrée [14,15]. Différents auteurs ont évoqué des facteurs exogènes dans la survenue de l’affection tels que des cicatrices de brûlure, de vaccination, de radiothérapie, des nævi traumatisés, des lésions syphilitiques, des microtraumatismes sur peau saine [9,16]. Dans notre série, aucun de ces facteurs n’a été retrouvé mais 10,2% des patients ont rapporté la notion de traumatisme antérieur. Cette notion est retrouvée dans 10 à 20% des cas selon les différentes séries [8,14,15]. Le retard diagnostique est comparable à celui observé dans les autres études pouvant atteindre 82 ans [2,7,9]. Le délai moyen séparant l’apparition de la lésion et la première demande de soins dans notre série est de 5,6 ans (un mois et 30 ans). Ce retard est expliqué par l’évolution lente et progressive de la lésion et l’absence de signes fonctionnels et de troubles généraux [9]. Dans notre série, six cas étaient des récidives (12%). Cette tendance à la récidive locale en l’absence d’une chirurgie adaptée est une donnée classique [7,17–19]. Cliniquement, le stade de début correspond à une plaque indurée, recouverte d’une peau d’aspect et de coloration normale, parfois brun rouge ou violacée. A un stade plus avancé (stade nodulaire), la plaque s’étale, sa surface devient irrégulière et bosselée, réalisant au bout de quelques mois à quelques années, une masse multinodulaire, souvent polychrome et dure. Cette évolution en deux stades n’est pas constante car certaines formes sont d’emblée uni-nodulaires ou multi-nodulaires [20]. Dans notre série, le DFS était le plus souvent nodulaire (59,2%). Non traitées, ces lésions peuvent devenir très volumineuses, ou bien s’ulcérer pour devenir douloureuses et hémorragiques [2,5,6,8]. L’ulcération était notée chez six patients et l’hémorragie dans trois cas. La lésion était douloureuse dans 24,5%. Ce taux est superposable aux données de la littérature où la douleur est notée dans 10 à 25% des cas [8].
Selon les publications, la tumeur mesure en moyenne un à cinq cm [8,9]. Dans notre série, la taille moyenne était de 4,7 cm. La taille maximale était de 20 cm, inférieure à celle de la série de Hammas (30 cm) [5]. Le DFS peut toucher n’importe quelle partie du corps avec une prédominance au niveau du tronc (surtout sa face antérieure) et des membres [9,13]. Selon les données de la littérature, l’atteinte du tronc est estimée entre 50 et 67% des cas [5,13]. Dans notre série, la topographie correspond aux données de la littérature avec une atteinte préférentielle du tronc dans 55,1% et des membres dans 26,5% des cas.
Comme dans notre série, l’état général des patients reste longtemps conservé [9]. Les métastases ganglionnaires et les métastases viscérales sont rares voire exceptionnelles et ne peuvent être retrouvées qu’après de longues évolutions ou une transformation sarcomateuse [9,16,17]. Elles n’ont été retrouvées chez aucun patient de notre série.
L’examen histologique est indispensable pour le diagnostic. La tumeur est faite d’une prolifération cellulaire dense, mal limitée, non encapsulée, occupant le derme, le plus souvent dans sa totalité. Elle envoie de fins prolongements parfois très profonds dans l’hypoderme, ce qui expliquerait la survenue de récidives même avec des marges de résection larges [5,9]. L’épiderme est respecté. Les cellules sont allongées, fusiformes, à cytoplasme plus ou moins abondant, à noyau ovalaire, régulier. Les mitoses sont variables avec de rares atypies. Le stroma est variable d’une zone à l’autre [5,9,16]. Les fibres collagènes et réticuliniques sont plus ou moins abondantes, tandis que les fibres élastiques sont refoulées à la périphérie de la tumeur. Au sein des amas de cellules néoplasiques, on distingue un nombre variable d’espaces vasculaires et des coulées cellulaires péri-nerveuses. Dans le temps survient une diminution progressive de la composante fibreuse conjonctive et une augmentation de la densité cellulaire. Sur le plan architectural, les cellules sont disposées en faisceaux rayonnants (aspect en “rayon de roue”) ou tourbillonnants. Quelques sous types histologiques particuliers et rares [14,15], méritent d’être mentionnés: la forme myxoide et la forme pigmentée ou tumeur de Bednar, retrouvées chacune dans un cas. Le DFS infiltre sans détruire et les annexes sont longtemps conservées. Il n’existe pas de réaction inflammatoire. Il s’agit d’une tumeur de faible grade de malignité [20]. En général, l’aspect histologique permet de guider le diagnostic. Dans les cas douteux, l’immunohistochimie permet de distinguer le DFS des autres tumeurs à cellules fusiformes. Elle montre une positivité intense et diffuse du CD34 et une négativité constante de la desmine et de la PS100 [4,9,21]. Les zones en transformation sarcomateuse n’expriment qu’exceptionnellement et de façon très faible le CD34 [9]. Les techniques de cytogénétique mettent en évidence deux types d’anomalies du caryotype à type de chromosome en anneau surnuméraire [17,22] ou une translocation [17,22]. Le bilan d’extension n’est recommandé que pour les patients dont l’examen clinique fait suspecter des métastases, en cas de DFS récurrent, ou en cas de transformation sarcomateuse. Ce bilan inclut une radiographie thoracique, une échographie abdominale et du trajet lymphatique [14].
Le traitement est difficile en raison de l’extension infra-clinique de la tumeur, pouvant être à l’origine d’une récidive. L’exérèse chirurgicale large est donc le traitement de référence [20]. Elle consiste à effectuer nécessairement des excisions larges et profondes en emportant une marge périphérique en peau saine de trois à cinq cm et en profondeur une barrière anatomique saine (13,20,22-26]. Il est évident que pour certaines localisations, telle que la face, cette marge de sécurité ne peut être respectée et devient même impossible. Dans ces localisations, la technique de Mohs mérite d’être discutée. Cette technique, faite avec de coupes horizontales, permet de réduire les marges à deux cm en moyenne, les marges étant réalisées sur mesure [27]. Elle a montré des taux de récurrence réduits avec une exérèse complète du DFS [21]. La reconstruction se fait par suture directe, cicatrisation dirigée ou par greffe cutanée ou lambeaux cutanés ou musculo-cutanés en fonction de la taille des lésions et leurs localisation. Le curage ganglionnaire systématique n’a aucun intérêt [8,13,16,28].
En raison de sa faible activité mitotique, le DFS n’est pas radiosensible [9,16]. La radiothérapie est préconisée dans les récidives multiples, les marges d’exérèse insuffisantes ou envahies, les tumeurs de très grande taille, les tumeurs primaires inopérables et les localisations empêchant une chirurgie large [13,14,26]. La chimiothérapie n’est pas une méthode efficace, toutefois, un certain espoir avec l’Imatinib (Glivec*) existe, et actuellement, plusieurs études cliniques sur le rôle néo adjuvent de cette molécule sont en cours [20,29].
Une surveillance clinique rigoureuse doit être maintenue, du fait de l’évolution lente et du haut pouvoir récidivant de cette tumeur [20]. Un suivi de trois à six mois est recommandé pour les trois à cinq premières années et un suivi annuel après [14,30]. Les récurrences après 5 ans peuvent se produire. Le suivi clinique peut être complété par une IRM dans quelques cas sélectionnés [14]. Le DFS est particulier par son agressivité locale et sa tendance aux récidives en l’absence de chirurgie adaptée. Ces dernières sont fortement corrélées avec des marges de résection incomplètes et sont estimées à 20% avec des extrêmes allant de 0 à 60% [20,21]. Les métastases sont beaucoup plus rares (5%) et sont pour la plupart pulmonaires [17,20]. La survenue de plusieurs récurrences est un facteur favorisant de cette dissémination [20] ainsi que la transformation sarcomateuse franchement maligne qui est exceptionnelle et qui se voit à un stade très tardif [5].
Les facteurs qui conditionnent le pronostic sont: la taille, la localisation au niveau de l’extrémité céphalique, une poussée évolutive rapide, des récidives rapides et surtout la qualité de l’exérèse initiale [2,5,13,20]. Celle-ci doit être radicale car elle constitue le facteur pronostique essentiel, conditionnant le risque de rechute locale [5,13].
CONCLUSION
Intermédiaire entre l’inoffensif fibrome et le redoutable sarcome, le DFS de DARIER FERRAND réalise une tumeur fibreuse de la peau d’un type particulier, rare, apparaissant à tout âge mais surtout à l’âge adulte. La localisation à la face antérieure du tronc est prédominante. Son évolution est locale sans troubles fonctionnels ni signes généraux. Le diagnostic, souvent tardif, est évoqué cliniquement et confirmé histologiquement, le recours à l’immunohistochimie se fait en cas de doute diagnostic. L’exérèse chirurgicale d’emblée large et profonde est le traitement de choix. Les thérapeutiques adjuvantes ne semblent avoir aucune place. Le pronostic vital étant rarement engagé par la prolifération, le pronostic est évalué sur le risque de récidive. Ainsi, le DFS est de bon pronostic lorsque le traitement est bien mené mais exige néanmoins une surveillance clinique longue.
Statement of Human and Animal Rights
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2008.
Statement of Informed Consent
Informed consent was obtained from all patients for being included in the study.
RÉFÉRENCES
1. Monnier D, Algros MP, Vidal MC, Danzon A, Pelletier F, Puzenat E, et al. Dermatofibrosarcome protubérant (tumeur de Darier et Ferrand):Etude épidémiologique rétrospective descriptive en Franche-Comtésur une période de 20 ans (1982- 2002). Ann Dermatol Venereol. 2005;132:607.
2. Joucdar S, Kismoune H, Boudjemia F, Bacha D, Abed, L. Les dermatofibrosrcomes de Darier et Ferrand. Analyse rétrospective de 81 cas sur dix ans (1983-1994). Ann Chirur Plast Esthét. 2001; 46:134-40.
3. Torreggiani WC, Al-Ismail K, Munk PL, Nicolaou S, O’Connell JX, Knowling MA. Dermatofibrosarcoma Protuberans:MR imaging features. Am J Roentgenol. 2002;178:989–93.
4. Nedelcu I, Costache DO, Costache RS, Nedelcu D, Nedelcu LE. Darier-Ferrand Dermatofibrosarcoma Protuberans with Peculiar Aspect. Balk Mil Med Rev. 2006;9:44–9.
5. Hammas N, Badioui I, Znati K, Benlemlih A, Chbani L, El Fatemi H, et al. Dermatofibrosarcoma protuberans:report of 27 cases and review of the literature. Pan Afr Med J. 2014;18:280.
6. Bendix-Hansen K, Myhre-Jensen O, Kaae S. Dermatofibrosarcoma protuberans:a clinico-pathological study of nineteen cases and review of world literature. Scand J Plast Reconstr Surg. 1983; 7:247 52.
7. TraoréSS, Zida M, Baro FT, Boukoungou G, Goumbri OM, Sano D, et al. Le dermatofibrosarcome de Darier et Ferrand (DFDF). Àpropos de 7 cas au CHU de Ouagadougou, Burkina Faso. Bull Soc Pathol Exot. 2007;100:105–6.
8. Taylor HB, Helwig EB. Dermatofibrosarcoma protuberans. A study of 115 cases. Cancer. 162;15:717–25.
9. Kasse A, Dieng M, Deme A. Les dermatofibrosarcomes de Darier et Ferrand àpropos de 22 cas et revue de la littérature. Med Afr Noire. 1999;46:222–7.
10. Moumine M, Armani A, Elkbabri M, Dandane MA, Elalami Z, Elmadhi T, et al. Dermatofibrosarcome de Darier et Ferrand congénital?:un cas pédiatrique. Rev Stomatol Chir Maxillofac. 2008; 109:393–5.
11. Marini M, Saponaro A, Magariños G, de Baldrich A, Lynch P, Remorino L. Congenital atrophic dermatofibrosarcoma protuberans. Int J Dermatol. 2001;40:448–50.
12. Pack GT, Tabah EJ. Dermato-fibrosarcoma protuberans. A report of 39 cases. AMA Arch Surg. 1951;62:391–411.
13. El Kacemi H, Aissa A, Bazine A, Kebdani T, Bougtab A, Benjaafar N. Dermatofibrosarcoma protuberans:report of 38 cases. Pan Afr Med J. 2014;19:274.
14. Acosta AE. CSV. Dermatofibrosarcoma Protuberans. Curr Treat Options in Oncol. 2017;96:1031–46.
15. Reha J, Katz SC. Dermatofibrosarcoma Protuberans. Surg Clin North Am. 2016;96:1031–46.
16. Elamrani D, Droussi H, Boukind S, Elatiqi K, Dlimi M, Benchamkha Y, et al. Dermatofibrosarcoma protuberans, particular skin tumor:report of 32 cases and review of the literature. Pan Afr Med J. 2014;19:196.
17. Hajji Ouafi FZ, Lamchaheb FE, Akazane A, Malzi I, Achachi L, El Fassy Fihri M, et al. Métastase pulmonaire d’un dermatofibrosarcome de Darier-Ferrand?:àpropos d’un cas. Ann Dermatol Venereol. 2011; 138:216.
18. Rowsell AR, Poole MD, M. Godfrey A. Dermatofibrosarcoma protuberans:the problems of surgical management. Br J Plast Surg. 1986;39:262–4.
19. Shmookler BM, Enzinger FM, Weiss SW. Giant cell fibroblastoma. A juvenile form of dermatofibrosarcoma protuberans. Cancer. 1989;64:2154–61.
20. Chouk Friaa S, Badri T, Hammami H, Benmously R, Marrak H, Debbiche A, et al. Dermatofibrosarcoma protuberans:a study of 18 cases. Tunis Med. 2013;91:342–345.
21. Shah KK, McHugh JB, Folpe AL, Patel RM. Dermatofibrosarcoma protuberans of distal extremities and acral sites:a clinicopathologic analysis of 27 cases. Am J Surg Pathol. 2017;00:1–7.
22. Bianchini L, Maire G, Pedeutour F. De la cytogénétique àla cytogénomique du dermatofibrosarcome de Darier-Ferrand (dermatofibrosarcoma protuberans) et des tumeurs apparentées. Bull Cancer. 2007;94:179–89.
23. Pack GT, Tabah EJ. Dermatofibrosarcoma Protuberans. AMA Arch Surg. 1951;62:391.
24. Vandeweyer E, Somerhausen NDSA, Gebhart M. Dermatofibrosarcoma Protuberans:How Wide is Wide in Surgical Excision??Acta Chir Belg. 2002;102:455–8.
25. Sun LM, Wang CJ, Huang CC, Leung SW, Chen HC, Fang FM, et al. Dermatofibrosarcoma protuberans:treatment results of 35 cases. Radiother Oncol. 2000;57:175–81.
26. Boujelbene N, Elloumi F, Hassine SB, Frikha M, Daoud J. Le dermatofibrosarcome de Darier et Ferrand:àpropos de 11 cas. Cancer/Radiothér. 2009;13:695–6.
27. Kostrzewa E, Beylot-Barry M, Vergier B, Pedeutour F, Beylot C. Dermatofibrosarcome atrophique multifocal àdébut pédiatrique. Ann Dermatol Venereol. 2006;133:359–61.
28. Das L, Grover SB, Chand K, Dawson L. Intracranial extension of a dermatofibrosarcoma protuberans of the scalp:a case report with brief review of literature. Surg Neurol. 2000;54:452–4.
29. Wright TI, Petersen JE. Treatment of Recurrent Dermatofibrosarcoma Protuberans with Imatinib Mesylate, Followed by Mohs Micrographic Surgery. Dermatologic Surg. 2007;33:741–4.
30. Tsai Y-J, Lin P-Y, Chew K-Y, Chiang Y-C. Dermatofibrosarcoma protuberans in children and adolescents:Clinical presentation, histology, treatment, and review of the literature. J Plast Reconstr Aesthetic Surg. 2014;67:1222–9.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
|

Comments are closed.