A phenotypic variant of Job-Buckley syndrome
Asmae Rasso , Hanane Baybay, Hakima Elmahi, Sara Elloudi, Fatima Zahra Mernissi
, Hanane Baybay, Hakima Elmahi, Sara Elloudi, Fatima Zahra Mernissi
Department of Dermatology, Hassan II University Hospital Center, Fes, Morocco
Corresponding author: Dr. Asmae Rasso, E-mail: rassoasmae@gmail.com
Submission: 18.02.2019; Acceptance: 12.05.2019
DOI: 10.7241/ourd.20194.18
Cite this article: Rasso A, Baybay H, Elmahi H, Elloudi S, Mernissi FZ. A phenotypic variant of Job-Buckley syndrome. Our Dermatol Online.
2019;10(4):382-384.
Citation tools:
BibTex | CSV | RIS | refer/BiblX | Endnote XML
Copyright information
© Our Dermatology Online 2019. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
The syndrome of hyper-immunoglobulinemia E or Job-Buckley syndrome is a rare primary immunodeficiency, due to a dysfunction of the STAT 3 gene. It is characterized by a triad associating a high immunoglobulin E (IgE), eczema and recurrent severe infections with staphylococcus. It’s about a patient of a 15-year-old t with a history of familial and personal atopy, a notion of bacterial and mycological infection repetitive since the age of 6 years without any similar case in the family. The clinical examination found a carapace of the scalp, a facial dysmorphism made of hypertelorism, epicanthus, mandibular prognathism, dental agenesis, macrocheilia, eczema, a stunting delay, a pubertal delay with an increase in total IgE. The patient was on oral antimycotic and topical, antibiotic, antihistamine, and vitamin C, with favorable evolution of more than 2 years. Buckley’s syndrome is a rare entity that should not be ignored before a table of recurrent skin infections, inflammatory dermatitis, and hyper-gammaglobulinemia E.
Key words: Hyperimmunoglobulinemia E, STAT 3 gene, Skin manifestations
INTRODUCTION
Autosomal dominant hyper-IgE syndrome (AD-HIES) described for the first time in 1966 by Davis et al. called «Job”. This syndrome was secondarily named by Buckley to a hyper-immunoglobulinemia E in 1972. It was in 1973 by Clark, then in 1974 when Hill showed that these two syndromes initially described rest on a defect of the chemotaxis of the neutrophils [1,2]. The physiopathology incriminates the mutation of the STAT3 gene. This gene is involved in the immune mechanisms. Indeed, the mutation of this gene will lead to an intensification of the production of immunoglobulin E by B lymphocytes, the loss of the modulation capacity of their production by IL 10 and IFN g [4]. This pathology affects a female subject, but especially both sexes are reached, without ethnic factor. The familial character has only rarely been described [1]. This syndrome is associated with particular clinical characteristics of immunological and morphological order [1–3]. The usual diagnostic criteria of SHIE include [9]: Hyper IGE greater than 2000 IU/ml, moderate hyper eosinophilia, eczematous dermatitis, recurrent bacterial infections, specially staphylococcus aureus, with a cutaneous seat, pulmonary, Ear Nose and Throat. We report Moroccan boy in a 16-year-old has a SHIE.
CASE REPORT
Our case was a boy with 16 years old, 6th of a sibling of 7, from a non-consanguineous marriage, well vaccinated, good psychomotor development until the age of 7 years without any similar case in his family, he had an atopy eczema, rhinitis, conjunctivitis, and asthma in family, he presented a several episodes of bacterial and mycological infection, at 7 years of age. He was hospitalized for management of a severe mycotic infection of the scalp. The clinical examination found facial dysmorphism with hypertelorism. epicanthus, mandibular prognathism and dental agenesis (Fig. 2), a macrocheilia (Fig. 2), a carapace of the scalp with adhering yellowish scales (Fig. 1), a stunted delay with – 5 (Deviation Standard) (Fig. 1), a pubertal delay P1, G2 according to TANNER classification. Ophthalmological examination revealed superinfected allergic conjunctivitis and the Ear, Nose and Throat examination showed chronical otitis media. The biological assessment revealed a hyper eosinophilia at 2950 element/ml, a biological inflammatory syndrome, the bacteriological and mycological scalp sampling were positive with staphylococcus and Trichophyton Rubran. viral, bacterial, parasitic serologies (Hepatitis B and C, HIV, CMV, syphilis toxoplasmosis) were negative. A level of immunoglobins. intradermal to tuberculin was negative, abdominal ultrasound had found the liver is dysmorphic with hypertrophy of the left lobe, regular contours and echo homogeneous structure, and homogeneous splenomegaly, trans-thoracic ultrasonography revealed left ventricular hypertrophy, and a conserved ejection fraction. The result of the skin biopsy is in favor of non-specific folliculitis. a biopsy of the salivary glands made objectified chronic Sialadenitis grade 1 of Shisolmet Masson without specificity. The diagnosis of job-Buckley syndrom was retained on clinical, biological and radiological criteria. The patient has benefited from a symptomatic treatment based on antiseptic antibiotic and antimycotic “Griseofulvin” without reponse and then “Terbinafine” in front of the non-improvement, then he has benefited from red and blue light for anti-inflammatory effect, antihistamine and emollient creams with good evolution. then he was treated by an antibiotic prophylaxis injection of “Penitard 600000 IU/3 weeks” associated with the trimethoprim-sulfamethoxazole and vitamin C, dental extractions. then application of topical corticosteroid subsequently after conversion of infectious balance with a very favorable evolution without relapse in more than 2 years (Figs. 3 – 5).
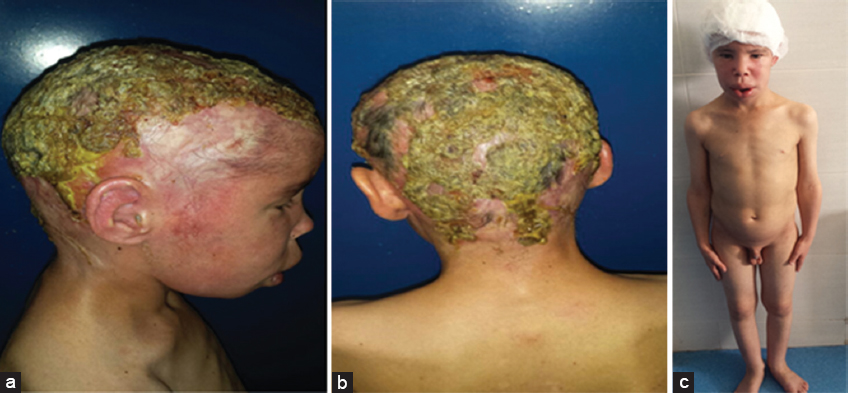 |
Figure 1: (a,b) Scalp carapasse, facial dysmorphia (c) a stunting and pubertal delay in 16 years old patient. |
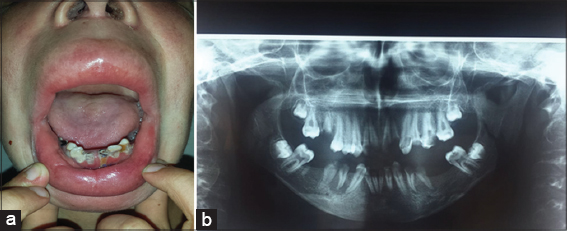 |
Figure 2: (a) Macrocheilia, (b) Mandibular prognathism and dental agenesis. |
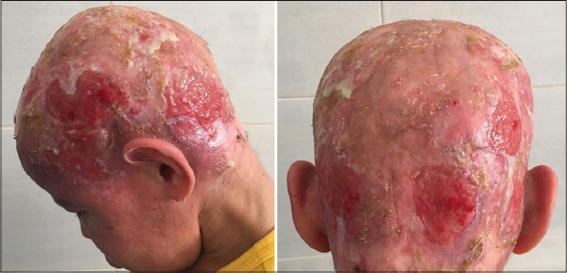 |
Figure 3: Favorable evolution after 6 months of treatment. |
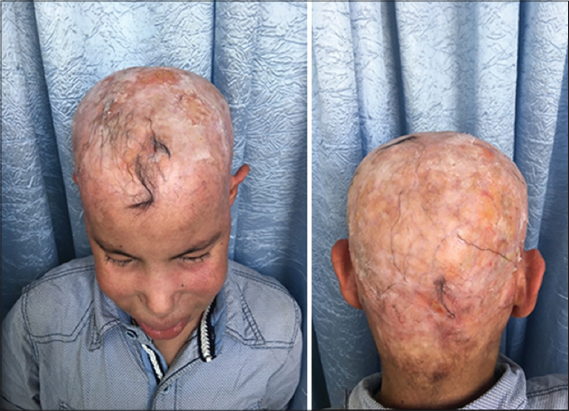 |
Figure 4: Favorable evolution after 2 years of treatment. |
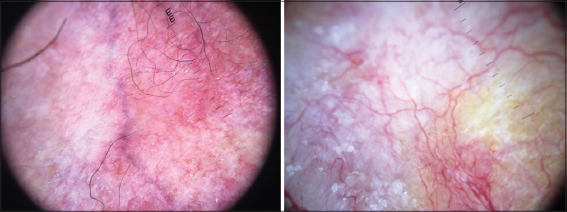 |
Figure 5: Cutaneous atrophy with telangiectasia at dermoscopy. |
DISCUSSION
Job-Buckley Syndrome or Hyper-Immunoglobulin E Syndrome (SHIE) is a rare multi-systemic infectious syndrome related to a primary immunological disorder. It affects both sexes, in the first weeks of life but sometimes at a later age as the case of our patient. This pathology is caused by the lack of chemotaxis of neutrophils [5] which leads to an immunodeficiency responsible for the generalized infectious manifestations characteristic of this syndrome [1,6]. These are bacterial and are found mainly in the skin as abscesses, pneumonia. The offending bacterial species are, staphylococcus aureus in 60% of cases, hemophilus influenzae in 10% of cases and, infrequently, streptoccoccus, pneumoniae, cryptococcus neoformans, aspergillus fumigatus [1,2]. Chronic candida infections, mainly related to Candida albicans [1]. This explains therecurence bacterial and mycological infections in our patient. From a morphological point of view, the manifestations are mostly facial asymmetry with hemi-hypertrophy, flattening of the base of the nose, prominent forehead [1,2,4,7], as reported in this case. They develop early in childhood. The manifestations are also oral with a delay of the loss of the temporary teeth in 75% of the cases causing a delay of eruption of the permanent teeth or an ectopic eruption of these teeth and malocclusions [8]. Mucosal lesions are often seen in the form of keratotic streaks or plaques that may be similar of lichenoid lesions [1]. Other inconsistent skeletal signs are reported, such as early cranial synostosis, osteoporosis and osteogenesis imperfecta causing frequent fractures of long bones, scoliosis [2]. On the dermatological level, this syndrome is characterized by the presence of a chronic dermatosis like an atopical dermatis preferentially evolving in the face, ears and scalp, and this, from the first months of life [1]. Other clinical and biological manifestations, but more inconsistent, are observed in this pathology such as vascular, skeletal, facial and oral abnormalities.
In early childhood, we can mention 2 differential diagnoses:
- High-dose atopic dermatitis of IgE with recurrent staphylococcal skin infections, for which a favorable evolution of IgE can be observed. However, the best criterion remains the absence of visceral involvement in dermatitis [6].
- Chronic granulomatosis must also be mentioned, it is frequently associated with osteomyelitis, whereas this is not observed in Job’s syndrome [6].
The treatment of Job-Buckley syndrome is purely symptomatic and concerns the infectious, dermatological and ocular manifestations. It is based on the regular and prophylactic use of antibiotics (including trimethoprim-sulfamethoxazole) The prescription of antibiotics, antihistamines and vitamins allows stabilization of lesions [6,9,10] as the case of our patient.
CONCLUSION
Job-Buckley syndrome is a rare immune disorder responsible for recurrent infections associated with high levels of serum IgE. There is currently no safe treatment that has clearly demonstrated efficacy. The prognosis depends primarily on the early management of pathology to reduce if possible, complications, particularly the infectious one.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Ikiss JA, Alaoui OM. Le syndrome de Job-Buckley:àpropos d’un cas et revue de la littérature. Med Buccale Chir Buccale. 2015;21:105-8.
2. DeWitt CA, Bishop AB, Buecher LS, Stone SP. Hyperimmunoglobulin E syndrome:two cases and a review of literature. J Am Acad Dermatol. 2006;54:855-65.
3. Buckley RH. The hyper IgE syndrome. Clin Rev Allergy Immunol. 2001;20:139-54.
4. Avery DT, Ma CS, Bryant VL, Santner-Nanan B, Nanan R, Wong M, et al. STAT3 is required for IL-21-induced secretion of IgE from human naive B cells. Blood 2008:1;112:1784-93.
5. Conti HR, Baker O, Freeman AF, Jang WS, Holland SM, Li RA, et al. New mechanism of oral immunity to mucosal candidias in hyper-IgE syndrome. Mucosal Immunol. 2011;4:448-55.
6. Dervaux B, Le Goff C, Turbanski F, Baidj Z, Coche R. Recurrent infection in a child caused by Job-Buckley syndrome. Arch Pediatr. 2009;16:122,142-5.
7. Borges WG, Hensley T, Carey JC, Petrak BA, Hill HR. The face of Job. J Pediatr 1998;133:303-5.
8. Bodard A-G, Delaunay T, Salino S. Manifestations buccales du syndrome de Job-Buckley: àpropos d’un cas. Med Buccale Chir Buccale. 2010;16:113-7.
9. Fégel P. Le syndrome du Buckley:Revue de la littérature et àpropos d’un cas. Thèse N°57. UniversitéHenri Poincaré, Nancy 1 2002.
10. Guerouaz N, Benhiba H, Hassam B. Le syndrome de Buckley:une nouvelle observation. Ann de Dermatol Vénéréol. 2013;140:S44-5.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
 http://orcid.org/0000-0002-7087-4411 http://orcid.org/000-0003-3455-3810 http://orcid.org/0000-0002-7087-4411 http://orcid.org/000-0003-3455-3810 |

Comments are closed.