EDSF (McGrath syndrome) – a rare variant of epidermolysis bullosa simplex
Kuldeep Verma, Reena K. Sharma, Mudita Gupta , Saru Thakur
, Saru Thakur
Department of Dermatology, Venereology and Leprosy, Indira Gandhi Medical College, Shimla, Himachal Pradesh, India
Corresponding author: Dr. Mudita Gupta, E-mail: muditadrgupta@yahoo.com
Submission: 01.09.2018; Acceptance: 22.01.2019
DOI: 10.7241/ourd.20193.15
Cite this article: Verma K, Sharma RK, Gupta M, Thakur S. EDSF (McGrath syndrome) – a rare variant of epidermolysis bullosa simplex. Our Dermatol Online. 2019;10(3):281-283.
Citation tools:
BibTex | CSV | RIS | refer/BiblX | Endnote XML | Wikipedia Citation Templates
Copyright information
© Our Dermatology Online 2019. No commercial re-use. See rights and permissions. Published by Our Dermatology Online.
ABSTRACT
Ectodermal Dysplasia Skin fragility syndrome (EDSF) is a rare autosomal recessive syndrome characterized by loss of function mutation in plakophilin 1 gene (PKP1). Around 20 cases of EDSF have been reported in literature with presentations of skin fragility, alopecia, palmoplantar keratoderma, hypohidrosis, nail dystrophy, cheilitis and few uncommon presentations. There are no diagnostic criteria for EDSF syndrome, patient with overlapping feature of ectodermal dysplasia and genetic blistering diseases are included in EDSF syndrome. We report a case of 11 months male infant admitted for pneumonia, with features of skin peeling over pressure sites since 2-3 months of age, along with sparse lustreless hair, absence of eyebrows and delayed dentition. The child was diagnosed as a case of EDSF syndrome.
Key words:McGrath, Ectodermal dysplasia, Skin fragility
INTRODUCTION
Ectodermal dysplasia-skin fragility (EDSF) syndrome also known as McGrath syndrome, is a rare autosomal recessive genodermatosis, which results from loss-of-function mutations in plakophilin 1 (PKP1) [1]. It is now considered as a specific suprabasal form of epidermolysis bullosa simplex [2]. First case of EDSF syndrome was described by Mcgrath in a 6 year old boy in 1997 [1]. There are only few case reports of EDSF syndrome, we are reporting this case to add to the number of cases of the rare entity.
CASE REPORT
Eleven months male infant admitted in paediatric department for pneumonia was referred to us with the complaints of peeling of skin at places and sparse hairs. On detailed interrogation, mother gave history of fluid filled lesions at the sites of friction since 2 months of age, lesions used to rupture spontaneously or with trivial trauma leaving behind raw surface which used to heal in 2-3 weeks time without any pigmentary changes or scarring. There was also history of sparse scalp hair, absence of eye brows and eyelashes. The child was born at term with normal vaginal delivery with birth weight- 2.6kg and unremarkable antenatal period. There was no history suggestive of lowered immunity. There was no history of consanguity, similar complaints in the family.
On examination, child had fever with increased respiratory rate and chest indrawing with other vitals normal and was on treatment for pneumonia. Child was weighing 6 kg and other milestones were normal.
On Mucocutaneous examination, there were multiple well to ill defined, erythematous irregular erosions of size approximately 2x2cm to 5x 4cm over back, legs and scalp (Fig. 1). Nikolsky’s sign was negative. Scalp hair were sparse and lustreless with absence of eyebrows and eyelashes (Fig. 2). Hair microscopy did not reveal any beading or shaft breakage. Teeth had not erupted and nails were lustreless. Biopsy was not done as the child was sick and parents were reluctant for biopsy.
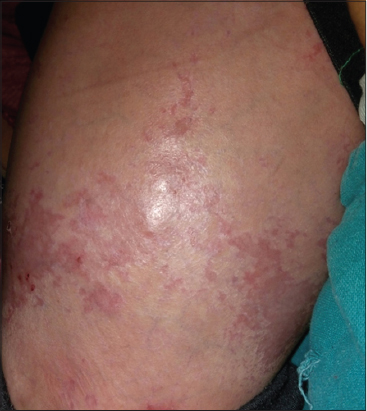
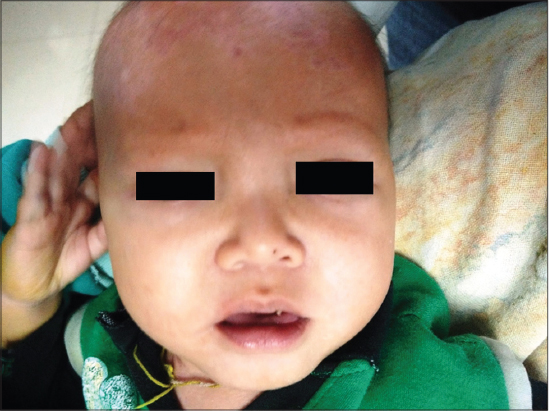
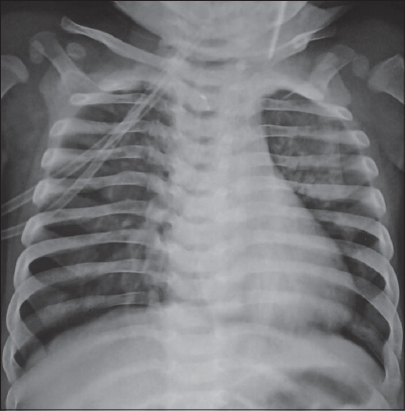
Systemic examination except respiratory system was normal. Lab investigations and chest radiography were suggestive of pneumonia (Fig. 3). On the basis of clinical and laboratory findings we diagnosed the case as ectodermal dysplasia-skin fragility syndrome.
DISCUSSION
Ectodermal dysplasias are a group of inherited disorder with developmental abnormalities of two or more of the following: hair, teeth, nails, sweat glands and other ectodermal structures. Genetic blistering disorders are a group of disorders characterized by blistering of the skin and mucosae following mild mechanical trauma. Very rarely, ectodermal dysplasia may co-exist with genetic blistering disorders, first case of which was described by McGrath in 1997 and coined the term “Ectodermal dysplasia-skin fragility syndrome” (EDSF) or McGrath syndrome [1].
EDSF is a rare autosomal recessive genodermatosis, which results from loss-of-function mutations in plakophilin 1 (PKP1), sequencing of genomic DNA revealed a homozygous 5 base pair deletion in exon 5 of the PKP1 gene [2]. PKP1 is member of the armadillo family of proteins which links desmosomal cadherins (desmogleins and desmocollins) to the keratins, contributing to the mechanical integrity of keratinocytes [3].
PKP1 is expressed throughout the epidermis, particularly in suprabasal cells and also in the outer root sheath of hair follicles [4]. Two isoforms of PKP1,1a and 1b have been described [5]. PKP1a is expressed in both desmosomes and nuclei, whereas PKP1b is expressed only in nuclei. Currently, the specific biologic significance of the two isoforms is unknown [4].
Most common features such as skin fragility, alopecia, palmoplantar keratoderma, hypohidrosis, nail dystrophy, and cheilitis, have been described in patients with EDSF syndrome. Uncommon features such as perioral fissuring, pruritus, growth retardation and dental caries reported in some cases of EDSF syndrome [5].
Unlike PKP 2 and other desmoplakins, PKP1 is not expressed in the heart so cardiac involvement is not seen in EDSF syndrome.
In Hay- wells syndrome patients present with alopecia, skin fragility along with cleft lip/palate and ankyloblephron, which was absent in our case. Mendes de costa syndrome also presents with skin blistering, alopecia, reticulate hyper/hypo pigmentation, but dentition is normal. No pigmentary changes were seen in our patient. A variant of monilithix with desmoglien 4 mutation also shows scalp erosions and congenital hypotrichosis, but absence of follicular papules and hypodontia in our patient favours EDSF syndrome.
Histopathological examination shows hyperkeratosis, acanthosis with widened intercellular spaces, and acantholytic keratinocytes [6]. Immunohistochemical and electron microscopic studies have demonstrated poorly developed, small desmosomes along with a reduction in the number of desmosomes in the epidermis, particularly involving the lower suprabasal layer [6].
Treatment options are generally limited in the form of counselling, skin care avoiding trauma, use of mild antiseptics. A prenatal diagnosis can be established by preimplantation genetic diagnosis [7].
As EDSF has manifestations of both epidermolysis bulosa simplex and ectodermal dysplasia, but all the clinical features described by McGrath may not be present in every case, especially at early age of presentation. Hopefully, more number of cases in future will help us establish diagnostic features for this syndrome.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. McGrath JA, McMillan JR, Shemanko CS, Runswick SK, Leigh IM, Lane EB, Mutations in the plakophilin 1 gene result in ectodermal dysplasia/skin fragility syndromeNat Genet 1997; 17: 240-4.
2. Alatas ET, Kara A, Kara M, Dogan G, Baysal O, Ectodermal dysplasia-skin fragility syndrome with a new mutationIndian J Dermatol Venereol Leprol 2017; 83: 476-9.
3. Adhe VS, Dongre AM, Khopkar US, Ectodermal dysplasia-skin fragility syndromeIndian J Dermatol Venereol Leprol 2011; 77: 503-6.
4. Kashyap S, Shanker V, Sharma N, Ectodermal dysplasia-skin fragility syndrome: A rare case reportIndian J Dermatol 2015; 60: 421-
5. McGrath JA, Mellerio JE, Ectodermal dysplasia-skin fragility syndromeDermatol Clin 2010; 28: 125-9.
6. Bergman R, Sprecher E, Histopathological and ultrastructural study of ectodermal dysplasia/skin fragility syndromeAm J Dermatopathol 2005; 27: 333-8.
7. Fassihi H, Grace J, Lashwood A, Whittock NV, Braude PR, Pickering SJ, Preimplantation genetic diagnosis of skin fragility–ectodermal dysplasia syndromeBr J Dermatol 2006; 154: 546-50.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Request permissions
If you wish to reuse any or all of this article please use the e-mail (brzezoo77@yahoo.com) to contact with publisher.
| Related Articles | Search Authors in |
|
|
|

Comments are closed.