|
Get Citation
|
|
|
Taj FT, BM V. Acquired partial lipodystrophy: Barraquer –Simons syndrome: A rare case report. Our Dermatol Online. 2019;10(1):59-61. |
|
|
Download citation file:
|
Acquired partial lipodystrophy: Barraquer –Simons syndrome: A rare case report
Farhana Tahseen Taj1, Vyshak BM2
1Department of Dermatology, Venereology & Leprosy, Jawaharlal Nehru Medical College, KLE Academy of Higher Education and Research, Belgaum – 590010, Karnataka, India, 2Post Graduate student (MD DVL), Department of Dermatology, Jawaharlal Nehru Medical College, KLE Academy of Higher Education and Research, Belgaum – 590010, Karnataka, India
Corresponding author: Dr. Farhana Tahseen Taj, E-mail: farhanahaveri@gmil.com
Submission: 21.05.2018; Acceptance: 28.07.2018
DOI:10.7241/ourd.20191.15
ABSTRACT
Acquired partial lipodystrophy (APL) or Barraquer-Simons syndrome is a rare form of progressive lipodystrophy of unknown etiology. Metabolic complications are less common than with other lipodystrophies. Patients usually have renal involvement in the form of membrenoproliferative glomeruloneprhitis which can lead to end stage renal disease. Here we report a case illustrating the importance of recognizing the clinical features of lipodystrophic syndrome, which may present with potentially severe consequences and psychological distress. A brief overview is made, addressing the disease, course, and its consequences.
Key words: Lipodystrophy, Barraquer simons syndrome, Membranoproliferative Glomerulonephritis
INTRODUCTION
Lipodystrophies are heterogenous group of disorders with selective loss of adipose tissue which can either be partial or complete. Barraquer Simons Syndrome is the first acquired partial lipodystrophic (APL) disorder described by Mitchell (1885), Barraquer (1907) and Simons (1911). It is also known as lipodystrophia progressive or Cephalothoracic lipodystrophy [1]. Gellis, Senior and Colleagues have described association of Acquired partial lipodystrophy (APL) with Membranoproliferative Gomerulonephritis (MPGN).
CASE REPORT
A 35 year old female presented to our skin OPD with the complaints of loss of fat over bilateral cheeks since 4 years. It was Insidious in onset which gradually progressed to involve bilateral arms, upper back & both the hips in a span of about 1 year. She also complained of increased loss of hair from the scalp and right eyebrow since 3 months. She was the second child of non-consanguineous, healthy parents. She had regular menstrual cycles. Her past medical history was insignificant and no intake of any drug. There was no history suggestive of recent viral infection, insulin resistance or any significant weight loss. There was no family history of the same condition.
On examination there was bilateral loss of buccal fat pads (Fig. 1) and prominent zygomatic arches. There was loss of subcutaneous fat over bilateral arms, (Fig. 2) upper back and bilateral waist. Furthermore, there were multiple nodular swellings, each measuring about 1×2 cm palpable in the submandibular region and solitary firm mass with ill-defined margins palpable in the lower inner quadrant of right breast. Scalp showed ill-defined areas of hair loss with thickened skin and loss of hair from the lateral aspect of right eye brow with thick underlying skin. The subcutaneous fat was preserved in other anatomic regions. Thyroid was normal on palpation. Hepatosplenomegaly, umbilical hernia, acanthosis nigricans, clitorimegaly, hirsutism or acromegalic features were absent. Systemic examinations were unremarkable, including her Ophthalmic and neurological status.
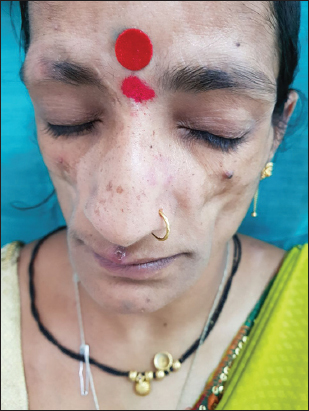
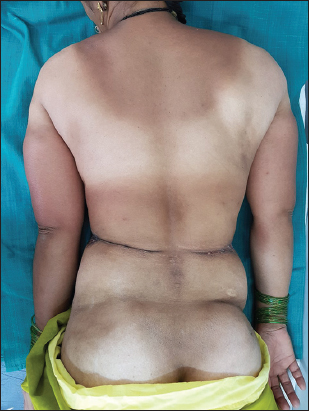
Her complete blood count, biochemical parameters (with renal and liver function tests), urine analysis with urinary albumin excretion, thyroid function tests revealed no abnormalities. Her fasting glucose and HbA1c presented normal values. There was reduced HDL-cholesterol with normal LDL cholesterol and triglycerides levels. The C3 levels were normal but towards lower level (94 mg/dL). ANA was negative with serum markers being nonreactive. Histopathology was suggestive of lipoatrophy (Fig. 3). Ultrasound of neck and breast suggested multiple lipomas and breast mass suggestive of BIRADS grade IV with moderate suspicion of malignancy respectively. Furthermore, Excision biopsy of breast mass reported chronic mastitis. The patient had neither renal involvement, nor any severe metabolic disorders until our last observation. She was counseled about the disease and was started on Tab Rosiglitazone 4mg OD and called for follow up.

DISCUSSION
There are four major categories of Lipodystrophies: Congenital generalized lipodystrophy (Berardinelli Seip syndrome), Familial partial Lipodystrophy, Acquired Generalised Lipodystrophy (Lawrence syndrome) and Acquired partial Lipodystrophy (APLD, Barraquer –Simmons syndrome. The new entity is HAART induced lipodystrophy in HIV patients [2].
Our patient had clinical features suggestive of APL fitting into essential diagnostic criteria proposed by Misra et al. (Table 1) and one of supportive criteria that is absence of family history. The etiology of APL is unclear. Most cases are sporadic with Female to Male ratio 4:1. Mutations in LMNB2 gene has been identified in few patients [3].

The onset of APL is in childhood or adolescence age group. Clinically it starts with fat loss in a cephelo-caudal distribution starting from face, neck, upper extremities and trunk sparing lower abdomen, thighs and legs [4]. The fat loss is because of Complement 3 Nephritic factor that blocks degradation of C3 Convertase enzyme and lysis of adipocytes that expresses factor D [5]. Approximately 20% of patients develop MPGN and some of these can progress to end stage renal disease requiring renal transplantation [6]. Metabolic complication is not seen in APL. Infections and autoimmune diseases have been linked with APL.
Our patient had essential criteria of fat loss in cephelocaudal distribution and supportive criteria absence of family history. Her metabolic profile was within normal limits. She had atypical presentation of loss of fat from waist; such presentation is unusual in APL.
Mangement of APL is diet and exercise. Metreleptin therapy is not useful in APL as serum leptin levels are not deranged. Thiazolidinediones has been tried in APL which improves insulin resistance and hyperglycemia and stimulates the growth of adipocytes [7,8]. Cosmetic procedures like autologous fat transfer, dermal fillers for fat loss has been tried.
CONCLUSION
APL is a very rare form of lipodystrophy. We report this case because of its atypical presentation and its importance of regular follow up and management to prevent metabolic and renal complications.
CONSENT
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Misra A, Peethambaram A, Garg A. Clinical features and Metabolic and Autoimmune derangements in Acquired Partial lipodustrophy. Report of 35 cases and Review of literature. Medicine. 2004;83:18-34.
2. Fiorenza CG, Chou SH, Mantzoros CS. Lipodystrophy:Pathophysiology and Advances in Treatment. Nat Rev Endocrinol. 2011;7:137-50.
3. Hegele RA, Cao H, Liv DM, Costain GA, Charlton-Menys V, Rodger NW, et al. Sequencing of the Reannotated LMNB2 Gene Reveals Novel Mutations in Patients with Acquired PaRTIAL Lipodystrophy. Am J Hum Genet. 2006;79:383-9.
4. Brown RJ, Chair C, Vilar DA, Chaung PT, Dunger D, Garg A, et al. The diagnosis and management of lipodystrophy syndrome:A Multi-Society Practice Guidelines. J Clin Endocrinol Metab. 2016;101:4500-11.
5. Garg A. Lipodystrophy:Genetic and Acquired Body Fat Disorders. J Clin Endocrinol Metab. 2011;96:3313-25.
6. Hussain I, Garg A. Lipodystrophy Syndromes. Dermatol Clin. 2008;26:569-81.
7. Oliveria J,FreitasP,Law E, Carvalho D. Barraquer Simons Syndrome:a rare form of acquired lipodystrophy. BMC Res Notes. 2016;9:175-9.
8. Parker VE,Semple RK. Genetic forms of Severe insulin resistance:what endocrinologists Should Know. Eur J Endocrinol. 2013;169:R71-R80.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.