|
Get Citation
|
|
|
Mansouri S, Mai S, Houssaini AS, Hassam B, Ismaili N. Primary leiomyosarcoma of the psoas in a patient with Neurofi bromatosis Type 1. Our Dermatol Online. 2018;9(3):296-298. |
|
|
Download citation file:
|
Primary leiomyosarcoma of the psoas in a patient with Neurofibromatosis type 1
Siham Mansouri, Sara Mai, Asmaa Sqalli Houssaini, Badr Hassam, Nadia Ismaili
Department of Dermatology, University Hospital IBN Sina Rabat, Morocco
Corresponding author: Dr. Siham Mansouri, E-mail: siham-mnsr@hotmail.fr
DOI:10.7241/ourd.20183.17
Submission: 30.11.2017; Acceptance: 24.01.2018
ABSTRACT
Neurofibromatosis Type 1 (NF1) is an autosomal dominant disorder that reduces the effectiveness of the neurofibromin tumor suppressor, resulting in an increased risk for benign and malignant soft tissue tumors. Leiomyosarcoma has been infrequently observed in NF1 patients. This case adds to the limited number of leiomyosarcomas reported in NF1 patients. This particular malignancy presented in a highly unusual location. A 15-year-old male with a previous diagnosis of NF1 presented a 3-month history of right-sided lumbago. Pelvic MRI revealed a large parenchymatous mass (69x66x115mm) in contact with the right psoas muscle. A biopsy guided by the scanner was performed. Histological examination with diffuse positivity of tumor cells for immunohistochemical muscle markers revealed high-grade leiomyosarcoma. A neoadjuvant chemotherapy was started based on Fluorouracil. The evolution was marked by the death of the patient 04 months later. To our knowledge, this is the first report of an NF1 patient who developed a primary leiomyosarcoma of the psoas. These rare tumors should be further evaluated. A careful follow-up of these patients is essential.
Key words: Neurofibromatosis type 1, Leiomyosarcoma, Psoas
INTRODUCTION
Neurofibromatosis Type 1 (NF1) is an autosomal dominant disorder that reduces the effectiveness of the neurofibromin tumor suppressor [1]. Mutations result in a predisposition to developing a variety of tumors of the central and peripheral nervous systems, as well as other malignancies. However, the occurrence of malignant tumors unrelated to the nervous system is rare [2]. Patients with NF1 can develop leiomyosarcoma less frequently than other malignancies. We report an NF1 patient who developed a leiomyosarcoma of the psoas. To our knowledge, this is the first report of an NF1 patient who developed a primary leiomyosarcoma of the psoas.
CASE REPORT
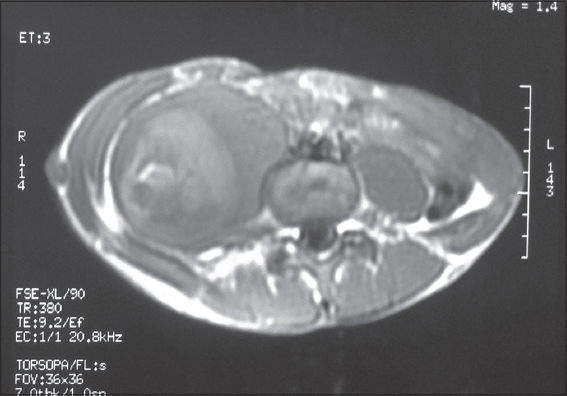
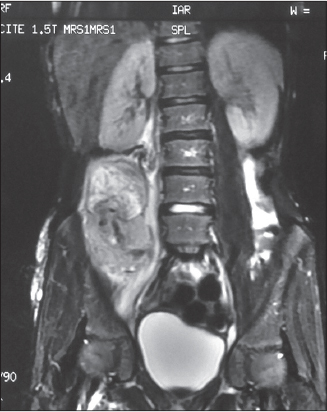
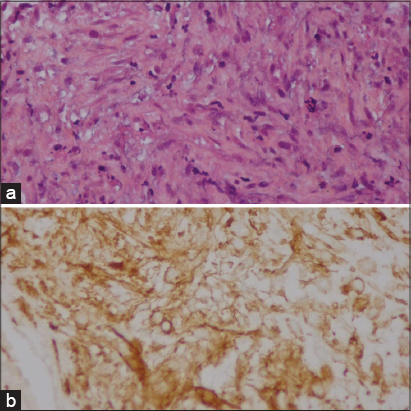
A 15-year-old male with a previous diagnosis of NF1 presented a 3-month history of right-sided lumbago. He had no history of recent trauma. On exam, the skin overlying the affected digit was intact; the palpation of the right flank was very painful. Routine blood tests and tumor markers in the blood were normal. Initial pelvic tomography revealed a soft tissue mass measuring 62x62x105 mm (Fig. 1). Pelvic MRI revealed a large parenchymatous mass (69x66x115mm) in contact with the right psoas muscle, showing inhomogeneous structure and containing necrotic areas (Fig. 2). A biopsy guided by the scanner was performed. Histological examination with diffuse positivity of tumor cells for immunohistochemical muscle markers (H-Caldesmon) revealed high-grade leiomyosarcoma (Fig. 3). The decision to omit the resection was made by the operating physician due to the specific location and the size of the lesion. A neoadjuvant chemotherapy was started based on Fluorouracil. The MRI monitoring showed an extension of the lesion despite 4 sessions of chemotherapy with a serious deterioration of the general condition of the patient. The evolution was marked by the death of the patient 04 months later. Prior to the study, patient gave written consent to the examination and biopsy after having been informed about the procedure.
DISCUSSION
Patients with NF1 are predisposed to the development of benign and malignant neoplasms, particularly those of neurogenic or neuroendocrine origin [2]. Zöller et al. reported a fourfold increase in the risk of developing a malignancy in patients with NF1 (24%); when compared to the general population [3].
Soft-tissue sarcomas represent about 8% of all malignant tumours of children and adolescents with NF1 [4]. Leiomyosarcomas arise from smooth muscle and represent 10-20% of all diagnosed sarcomas [5]. Leiomyosarcoma is uncommon in NF1. Zöller found two cases of leiomyosarcoma among 70 patients with NF1 [3]. The different localizations of leiomyosarcomas reported in NF1 patients are [6]: the sciatic nerve, the liver, the bladder, the pelvis, intracranial and the hand [7]. Low-grade malignant peripheral nerve sheath tumor with smooth muscle differentiation has also been reported in the literature [6]. The Leiomyosarcoma of the psoas in NF1 has never been reported. This different localizations illustrates the need to be aware of potential leiomyosarcoma in NF1.
Leiomyosarcomas are often unresponsive to chemotherapy and radiation; thus, wide margin surgical removal has proven to be the most accepted treatment method [8]. New approaches to the treatment of these rare malignancies and the genetic interaction of the NF1 gene should be taken into consideration. A careful follow-up of these patients is essential.
CONCLUSION
This study adds to the limited number of cases of leiomyosarcomas that have been reported in NF1 patients. As NF1 patients may die at a young age after the onset of an associated malignancy, it is important to examine routinely these patients in order to have an early diagnosis and treatment for a better outcome.
CONSENT
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Bikowska-Opalach B, Jackowska T. Neurofibromatosis type 1 – description of clinical features and molecular mechanism of the disease. Med Wieku Rozwoj. 2013;17:334-40.
2. Andonaba J-B, Some OR, Sanon BG, Diallo B. Léiomyosarcomes cutanés primitifs révélés par une récidive tumorale des tissus mous [Primary cutaneous leiomyosarcoma revealed by soft tissue tumor recurrence]. Our Dermatol Online. 2017;8:449-52.
3. Zöller ME, Rembeck B, Odén A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer 1997;79:2125–31.
4. Bien E, Stachowicz-Stencel T, Sierota D, Polczynska K, Szolkiewicz A, Stefanowicz J, et al. Sarcomas in children with neurofibromatosis type 1-poor prognosis despite aggressive combined therapy in four patients treated in a single oncologic
5. Serrano C, George S. Leiomyosarcoma. Hematol Oncol Clin North Am. 2013;27:957-74.
6. Afşar CU, Kara IO, Kozat BK, Demiryürek H, Duman BB, Doran F. Neurofibromatosis type 1, gastrointestinal stromal tumor, leiomyosarcoma and osteosarcoma:Four cases of rare tumors and a review of the literature. Crit Rev Oncol Hematol. 2013;86:191-9.
7. Desai VS, Kakazu R, Crawford AH, Sheil A, Sorger J. Primary Leiomyosarcoma of the Hand in a Patient with Neurofibromatosis Type 1. Am J Cancer Case Rep. 2016;4:145-51.
8. Angelini A, Barastegui D, Gambarotti M, Ruggieri P. Leiomyosarcoma of the hand. Handchir Mikrochir Plast Chir. 2015;47:139-41.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.