|
Get Citation
|
|
|
Taj FT, Vupperla D, Raichur S, Ardeshana J. Werner’s syndrome: A case report and review of literature. Our Dermatol Online. 2018;9(2):148-151 |
|
|
Download citation file:
|
Werner’s syndrome: A case report and review of literature
Farhana Tahseen Taj, Divya Vupperla, Sridevi Raichur, Jolika Ardeshana
Department of Dermatology, JNMC Medical College and Hospital, Belagavi, India
Corresponding author: Dr. Farhana Tahseen Taj, E-mail: farhanahaveri@gmail.com
Submission: 30.08.2017; Acceptance: 25.11.2017
DOI: 10.7241/ourd.20182.10
ABSTRACT
Werner’s Syndrome also known as Pangeria is an autosomal recessive disorder characterized by premature aging, increased risk of malignancies and atherosclerosis. The Global incidence rate is less than 1 in 100,000 live births. The incidence is higher in Japan and Sardinia affecting 1 in 20,000 – 40,000 live births and 1 in 50,000 live births respectively. Individuals with this disorder typically grow and develop normally until they reach puberty. Affected individual usually do not have a growth spurt, resulting in short stature. The characteristic aged appearance typically begins to develop when they are in their twenties and includes graying and loss of hair, a hoarse voice, and thin, hardened skin. They may also have a facial appearance described as “bird-like facies.” Werner’s Syndrome has been described as “Caricature of ageing” by Epstein et al. 1996. An OMIM number # has been assigned to Werner’s Syndrome with phenotype MIM number 277700 (OMIM#277700).
Key words: Pangeria, Premature ageing syndrome, Bird like facies, OMIM277700
INTRODUCTION
Werner’s Syndrome (WS) is an autosomal recessive disorder affecting the connective tissue of the whole body. It is also known as Progeria adultorum and Pangeria. Werner’s Syndrome is considered one of the genomic instability syndromes. It is characterized by short stature, senile appearance, cataracts, joint contractures, early menopause, and premature arteriosclerosis, Scleroderma like features, premature canities, baldness, ulceration and increased risk of malignancy.
The highest incidence of WS reported is in Japanese patients (1000 of 1300 cases reported worldwide). We report a 33 year old male patient presenting to our outpatient department with history of keratosis over both the feet.
CASE REPORT
A 33 year old unmarried male presented with history of painful lesions over pressure bearing areas of both the feet since 5 years. Patient gives history of hoarseness of voice since 6 months. He had undergone surgery for cataract 10 years back. Patient had shown to a surgeon where he had advised a Doppler study for both the limbs one month back. The Doppler study showed early arteriosclerotic changes of both lower limbs. Venous system was normal.
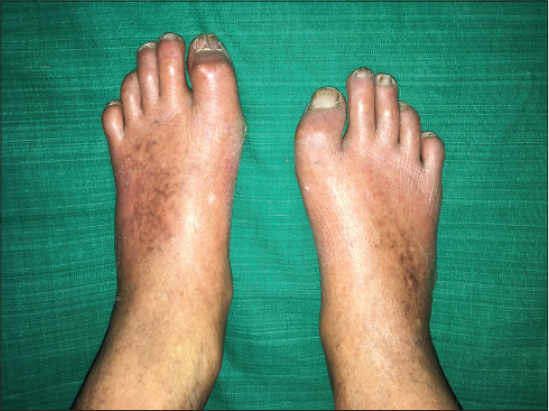
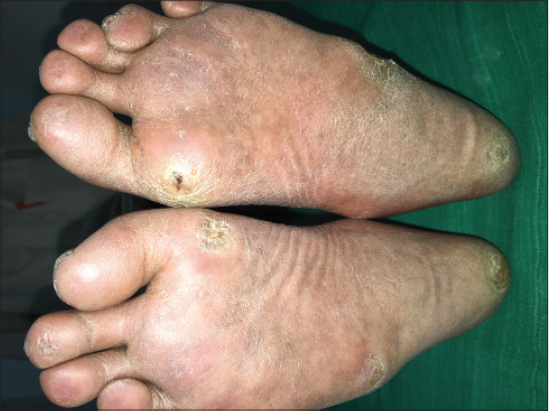
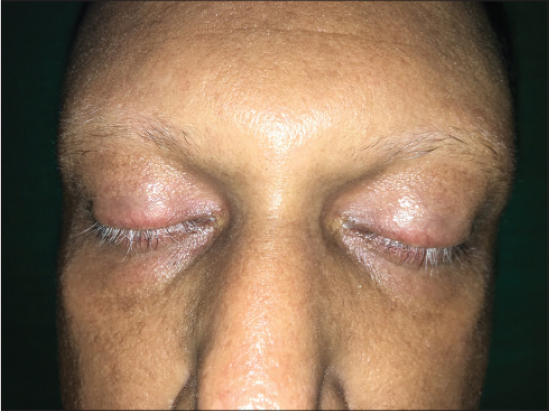
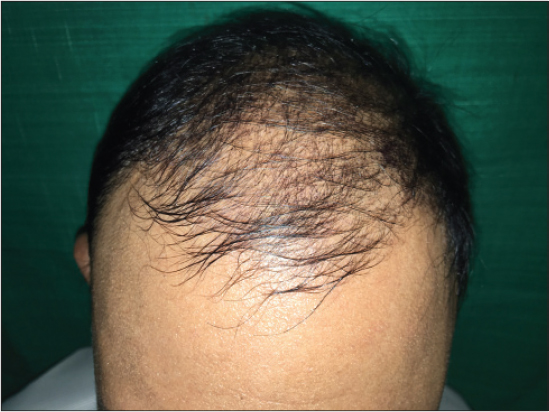
Patient was born of consanguineous marriage. He has one female sibling affected with similar disease. On physical examination the skin over both the limbs were shiny, with mottled pigmentation couldn’t pinched off (Fig. 1). Multiple hyperkeratotic lesions were seen over pressure bearing areas of both the limbs (Fig. 2). Contractures were seen over toes of both the feet but more on left feet. The trunk was normal. There was premature greying of eyelashes (Fig. 3) with receding scalp hair (Fig. 4). Biopsy was taken from the skin over left leg.
Histopathology showed sparse superficial perivascular and periappendageal lymphohistiocytes with occasional plasma cells. There is marked thickening of collagen bundles in upper reticular and papillary dermis. The thickened bundles are closely packed so as to give hyalinised appearance. The upper dermis shows telangiectasia.
DISCUSSION
Werner’s syndrome was first described by Otto Werner at 1904 [1]. He reported four cases of brothers and sisters, where he observed juvenile cataract, Pachyderm like alteration of the extremities, small stature, premature ageing of the face, juvenile grey hair and genital hypoplasia. In 1934, Oppenheimer and Kugel [2] described additional endocrine abnormalities such as osteoporosis and type 2 diabetes mellitus. The diagnostic criteria proposed for Werner syndrome by International registry for Werner syndrome 2000- 2005 given in Table 1.

Werner’s syndrome is a genetic disorder transmitted in an autosomal recessive pattern that affects males and females in equal numbers. The disorder frequency has been estimated at 1 to 20 per one million individuals in the Unites States. In the Japanese population it is 1 per 20,000 to 1 per 40000.
Werner Syndrome is caused by null mutations of WRN gene located on the short arm of chromosome 8 (8p12- 11.2) [3]. More than 80 different mutation of WRN gene have been identified. WRN gene codes for a member of Rec Q family of DNA helicases [7]. The disease is associated with abnormal metabolism of connective tissue. There is excessive synthesis of collagen I and III which is dependent on increased mRNA levels. Fibroblasts exhibit genomic. Instability, increased sensitivity to DNA damaging agent, lengthened S phase, accelerated replicative senescence.
Chromosomal instability including dysfunction of telomere maintainance is more prominent in WS cell than in normal cell. The accumulation of DNA double strands breaks (DNBs) [3] at G1 phase including those at telomere foci is accelerated in WS cells even at a low senescence level. These results indicate that WS cells are prone to accumulate DSBs spontaneously due to defect of WRN gene which leads to increased chromosomal instability that could activate check points resulting in accelerated senescence. The accelerating ageing in WS is due to increased levels of inflammatory cytokines produced by senescent Cells. The sclerodermatous skin and blood vessel wall calcification contributes to the development of non-healing ulcers and keratosis of the limbs. Individuals with WS develop normally till puberty. The onset can be in the mid- term but can be seen even after the age of 30 years. Patient with WS show increased risk for the development of malignant lesions and malignancy is a frequent complication of WS. The frequency of malignant lesion reported ranges from 5.6% to 25%. For general population epithelial cancer has an incidence of 10 times that of mesenchymal sarcoma, but among patients with Werner Syndrome incidence is approximately equal [5]. An increased risk of thyroid cancer, malignant melanoma, osteosarcoma and soft tissue sarcoma is also noted. Defective homologous recombination is believed to be the primary reason for chromosome abnormalities and genomic instability thus causing greatly increased risk of cancer. The severity of WS is due to its several complications dominated by atherosclerosis. Patients may develop different forms of atherosclerosis specially that affecting the coronary arteries leading to myocardial infarction that is the first cause of death in WS [4]. Increase in the blood hyaluronic acid levels is responsible for sclerodermatous changes and cardiovascular abnormalities [6].
The clinical manifestation starts with greying at the temples as early as 14 to 18 years of age. The skin manifestations are usually noticed at 18 and 30 years of age. The sclerodermatous skin changes gives rise to thin spindle shaped limbs contrast with the normal trunk. The facial appearance shows beaking of nose with bird like facies, shallow orbits giving rise to proptosis. The joints become fixed; with scelordactyly and acral gangrene can be present. Keratosis over pressure points on the feet and ankle separate to leave indolent ulcers. The voice may be high pitched and hoarse because of thickening of vocal cords. Most patient are of short stature and hypogonadal, some may achieve normal stature and successful pregnancies. Diabetes is seen in atleast 30% of patients. Cataracts are seen at the age of 20 and 38 years and mostly of posterior and subcapsular cataracts [8]. The radiological changes show calcification of arteries, ligaments, tendons and subcutaneous tissue with osteoporosis of the legs.
The differential diagnosis include other premature aging syndromes like Progeria, Acrogeria, Rothmund-Thomson Syndrome, Cockayne syndrome, hypohidrotic ectodermal dysplasia and Huriez Syndrome. Progeria is a rare condition with onset as early as 2 years of age. Affected individual shows short stature, large bald head with prominent veins, prominent eyes with bird like facies, coxa valga. Acrogeria is characterized by cutaneous atrophy and loss of subcutaneous fat particularly over distal extremities. Micrognathia, hollow cheeked and owl- eyed appearance. The affected individual has normal stature, normal life expectancy and scalp hair. Cockayne Syndrome is characterized by photosensitivity, Mickey Mouse facies, and normal scalp hair with disproportionately large extremities. In Rothmund Thomson syndrome the features are atrophy, telangeiectasia and mottled pigmentation most intense on sun exposes areas scalp hair is absent. Bilateral cataracts have developed between fourth and seventh year of life. Hypohidrotic ectodermal dysplasia is another differential where conical teeth, with reduced and absent sweating is seen. Huriez syndrome is an autosomal dominant transgrediant keratoderma characterized by keratoderma with scleroatrophy and nail changes.
Our patient had history of cataract surgery at the age of 20 years. The skin over the lower extremities was thin shiny with keratosis on pressure points. There was hoarseness of voice with proptosis of eyes and receding scalp hair. Doppler showed atherosclerotic changes of arteries of lower limb. These findings led us to make the diagnosis of premature aging syndrome. The histopathology showed thickening of collagen bundles with hyalinization just like sclerodermatous changes. Excluding all other premature aging syndrome considering his clinical features a diagnosis of Werner syndrome was made. Treatment given for the patient was mainly symptomatic. The patient has been referred to cardiologist and endocrinologist for further management.
REFERENCES
1. Werner CW. Uber Katarakt in verbindung mit Sklerodermie [Doctoral dissertation] Kiel, Germany:Kiel University, Schmidt &Klaunig;1904.
2. Kugel VH, Oppenheimer BS. Werner’s syndrome:report of the first necropsy and findings in a new case. Am J Med Scien. 1941;202:629-42.
3. Ariyoshi K, Suzuki K, Goto M. Watanabe, and S. Kodama, “Increased chromosome instability and accumulation of DNA double strand breaks in Werner syndrome cells. J Rad Res. 2007;48:219-31.
4. Ajili F, Garbouj W, Boussetta N, Laabidi J, Abdelhafidh NB, Louzir B, et al. Werner Syndrome:A new case report. Our Dermatol Online. 2013;4:490-2.
5. Yamamoto K, Imskiire A, Miyagawa N, Kashara T. A report of two cases of Werner’s syndrome and review of the literature. J Orthop Surg (Hong Kong). 2003;11:224-33.
6. Sert M, Fakioglu K, Tetiker T. Review of Two Siblings with Werner’s Syndrome:A Case Report. Case Rep Med. 2009;2009:138312.
7. Khan GA, Azfar NA, Malik LM, Sammar T, Jahamgir M. Werner syndrome:A case report and review of literature. J Pak Assoc Dermatol. 2011;21:304-8.
8. Burrow NP, Lovell CR. Disdors of connective tissue. In:Burns sT, Breathnach S, Cox N, Griffiths C, eds. Rooks’s Textbook of Dermatology, 8th edition. London:Blackwell Science;2010. 45.57-45.58.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.