|
Get Citation
|
|
|
Alouani I, Bkiyar H, Dikhaye S, Housni B, Zizi N. Overlapping of Sturge Weber syndrome and Klippel Trenaunay syndrome: A new case report. Our Dermatol Online. 2018;9(1):101-102. |
|
|
Download citation file:
|
Overlapping of Sturge Weber syndrome and Klippel Trenaunay syndrome: A new case report
Imane Alouani1, Houssam Bkiyar2, Siham Dikhaye1, Brahim Housni2, Nada Zizi1
1Department of Dermatology, Mohammed 6 University Hospital of Oujda – Medical School of Oujda, Mohammed First University of Oujda, Morocco; 2Department of Intensive Care, Mohammed 6 University Hospital of Oujda – Medical School of Oujda, Mohammed First University of Oujda, Morocco
Corresponding author: Dr. Imane Alouani, M.Sc., E-mail: alouani01@gmail.com
Submission: 05.06.2017; Acceptance: 10.06.2017
DOI: 10.7241/ourd.20181.33
Sir,
Sturge Weber syndrome (SWS) is a mesodermal phakomatosis characterized by meningo-facial angiomas with cerebral calcification. Klippel Trenaunay syndrome (KTS) is another very rare type of phakomatosis with cutaneous angiomas, varicose veins and enlargement of soft tissue or bones. Overlap between SWS and KTS is very rarely encountered. We report a case of 4-year-old girl with overlapping features of both SWS and KTS.
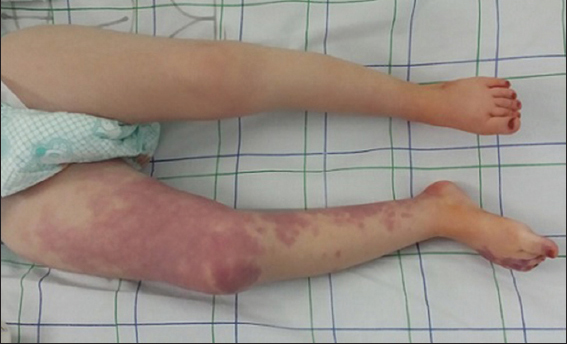
We report the case of a 4-year-old female patient with history of epilepsy and development delay, who presented a complex congenital neurocutaneous syndrome with symmetrical port-wine stain of the face, neck, trunk and arms (Fig. 1). There was also a hemangioma of the right thigh, knee and leg with thigh hypertrophy (Fig. 2). Computerizing tomography showed cerebral atrophy with bilateral leptomeningeal calcifications (Fig. 3).
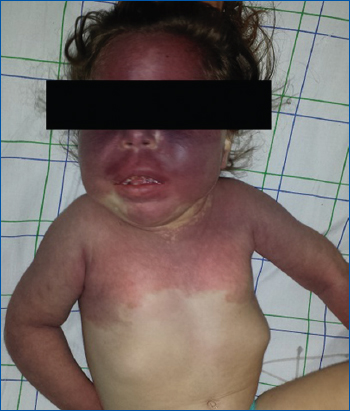
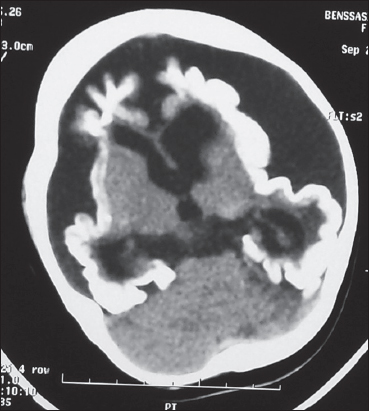
In our case, the patient presented congenital vascular malformations with port wine hemangioma and leptomeningeal calcifications, leading to a diagnosis of SWS. Known also as Sturge-Weber-Krabbe syndrome or Sturge-Weber-Dimitri, it is neuro-oculo-cutaneous syndrome classically associated with port wine stain in the ophthalmic division of the trigeminal nerve, ipsilateral occipital leptomeningeal angiomata, and inconstant glaucoma and vascular eye abnormalities. It was first described by Sturge in 1879 in a 6-years-old child with epilepsy, facial hemangioma and glaucoma. Weber reported in 1992 cerebral calcifications in patients presenting the same symptomatology [1]. The bilateral and extensive presentation of the hemangioma and neurological involvement as in our case is unusual. It is correlated to poor prognosis. Even though the ocular involvement is a part of the definition of SWS, it is not found in 65% of cases [2].
On the other hand, the patient presented a limb hypertrophy with hemangioma, which yielded a diagnosis of another vascular syndrome: KTS. Diagnosis criteria are: extensive cutaneous vascular malformation of a limb, congenital or acquired varicosities of the same limb and tissue or bone hypertrophy of the same limb [3]. Its incidence is about 1:100.000.
During the last 30 years, there have been more than 40 sporadic case reports of association of these two syndromes suggesting an overlap syndrome with genetic abnormalities. Pietruschka suggested that the SWS and KTS are different expressions of the same disease [4]. Sharma agreed with this hypothesis and suggested that the syndromes should be named neurocutaneous angioma [5].
Recently, Happle postulated a concept for the genetic basis of sporadic congenital abnormalities with capillary malformations, the “lethal gene” theory [6]. The phenomenon that one organism is composed of genetically different cell populations derived from a genetically homogeneous zygote is called mosaicism. These sporadic congenital abnormalities are not hereditary and the cutaneous lesions are distributed in a mosaic pattern usually following the lines of Blaschko [6,7].
Our observation shows again the phenotypic complexity of phakomatoses. These syndromes can result with severe neurological and vascular complications.
REFERENCES
1. Maruani A. Syndrome de Sturge-Weber. Presse Med. 2010;39:482-6.
2. Comi A. Update on Sturge-Weber syndrome:diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-64.
3. Verhelst H, Coster RV. Neuroradiologic findings in a young patient with characteristics of Sturge-Weber syndrome and Klippel-Trenaunay syndrome. J Child Neurol. 2005;20:911-3.
4. Pietruschka G. Zur Symptomatik der Syndrome nach Sturge-Weber and Klippel-Trenaunay. Klin Monatsbl Augenheilkd 1960;137:545–57.
5. Sharma P, Arya AV, Azad RV. Unusual retinal manifestation in a combination of Sturge-Weber and Klipplel-Trenaunay syndrome –a case report. Indian J Ophthalmol. 1990;38:195–7.
6. Happle R. Lethal genes surviving by mosaicism:a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899-906.
7. Vissers W, Van Steensel M, Steijlen P, Renier W, Van De Kerkhof P, Van Der Vleuten C. Klippel-Trenaunay syndrome and Sturge-Weber syndrome:variations on a theme? Eur J Dermatol. 2003;13:238–41.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.