|
Get Citation
|
|
|
Tiwary AK, Mishra DK, Chaudhary SS. A rare case of familial reactive perforating collagenosis. Our Dermatol Online. 2017;8(2):170-173. |
|
|
Download citation file:
|
A rare case of familial reactive perforating collagenosis
Anup Kumar Tiwary, Dharmendra Kumar Mishra, Shyam Sundar Chaudhary
Department of Dermatology, Venereology and Leprosy, Rajendra Institute of Medical Sciences, Ranchi, India
Corresponding author: Dr. Anup Kumar Tiwary, E-mail: anup07tunnu07@gmail.com
Submission: 28.03.2016; Acceptance: 01.10.2016
DOI: 10.7241/ourd.20172.45
ABSTRACT
Reactive perforating collagenosis is a rare disorder of transepidermal elimination in which genetically altered collagen is extruded out through the epidermis. This disease usually starts as asymptomatic or pruritic umbilicated papules on extensor aspects of extremities and face, and the lesions become more conspicuous with age. It has two forms acquired and inherited form, of which inherited form is extremely rare. Here, we document a case of inherited form of reactive perforating collagenosis in a 13 year old boy having multiple asymptomatic hyperpigmented umbilicated papules with a central keratotic plug distributed chiefly over face and extensor aspect of upper and lower extremities since his infancy, with history of similar lesions in his 18 year old elder brother.
Key words: Reactive perforating collagenosis; Transepidermal elimination; Umbilicated
INTRODUCTION
Reactive perforating collagenosis (RPC) is a rare skin disorder characterized by transepidermal elimination (TEE) of altered collagen through the epidermis [1–3]. Two distinct forms of RPC are known: the rare inherited form that starts in early childhood, usually precipitated by environmental cold or trauma; and the commonly acquired form that occurs in adulthood [2,4]. To the best of my knowledge, less than 50 cases of the inherited form of RPC are reported in the literature [5]. We herein report a rare case of inherited form of RPC in a 13 year old child, with history of similar lesions in his elder brother.
CASE REPORT
A 13 year old boy came to our outpatient department presenting with complaints of multiple, mildly pruritic, hyperpigmented papular and nodular lesions on face, extensor aspect of upper and lower limbs since his infancy. There was history of exacerbation and spontaneous remission but specific aggravating or relieving factors could not be found. During infancy, lesions started developing as skin coloured papules which followed the typical course of evolution progressing with increase in pigmentation and size of the lesions and later showing central umbilication and keratotic plug formation in each lesion. There were tendency in some lesions to heal spontaneously within 2 months of development leaving behind residual hyperpigmentation. Oozing, vesiculation or crusting were absent in most of the lesions. There was history of similar complaints in his 18 years old elder brother since his early childhood. History of parental consanguinity was present suggesting the possibility of autosomal recessive mode of inheritance in this familial case because none of the parents were affected with this disorder.
Local cutaneous examination demonstrated multiple, hyperpigmented, well defined, discrete, hyperkeratotic papulonodular lesions over face (on forehead and cheeks), both elbows, wrists, dorsa of hands, anterior aspect of both legs and feet (Figs. 1–4). The characteristic adherent central keratotic plugs were seen in most of the lesions because lesions were in different stages of evolution. Koebnerization due to scratching were also noted in some of the lesions. No hair structure was seen within any of the invaginations. Examination of nails, hair, teeth, and mucosa were normal and general physical and systemic examination was not remarkable. All routine laboratory parameters including urine analysis for albumin and microscopy were within normal limits.
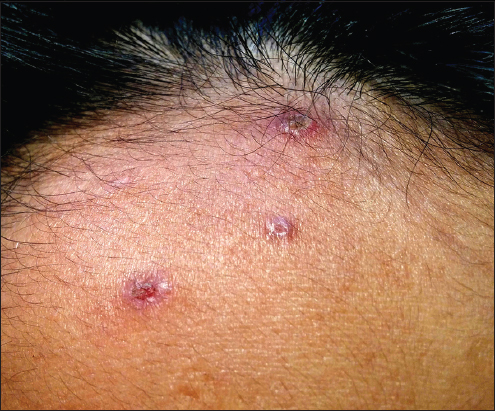
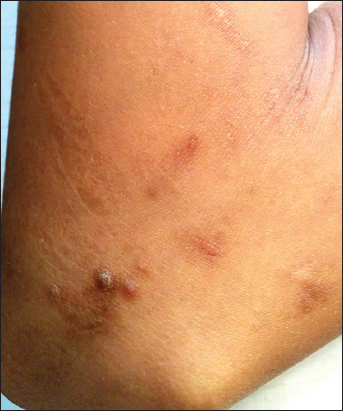
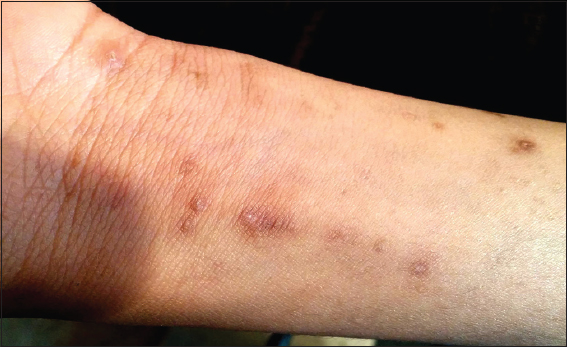

To come arrive at a conclusive diagnosis of specific perforating dermatosis, a punch biopsy sample was taken from the classical lesion over wrist and sent for histopathological evaluation (HPE).
HPE revealed epidermal invaginations filled up with orthokeratotic corneocytes, pyknotic neutrophils, and basophilic necrotic debris suggestive of altered collagen bundles. The invagination in its center showed break in continuity of epithelium. The dermis beneath the invagination had moderately dense infiltrate of lymphocytes and neutrophils. The epidermis around invagination showed moderate psoriasiform hyperplasia, acanthosis, mild spongiosis and focal parakeratosis (Fig. 5).
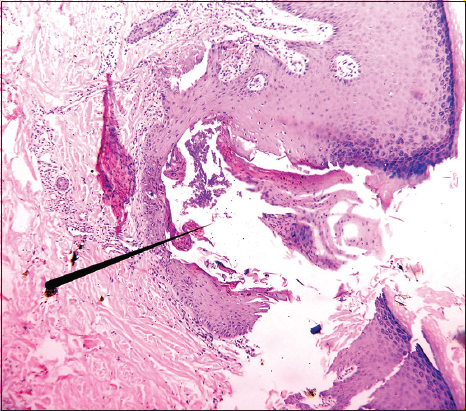
Based on clinical and histopathological features, a diagnosis of reactive perforating collagenosis was made. For management emollient, tretinoin cream and oral antihistaminics were given as first line of treatment and patient was advised to follow up on monthly basis.
Prior to the study, patient gave written consent to the examination and biopsy after having been informed about the procedure.
DISCUSSION
Perforating dermatoses are a group of uncommon skin disorders characterized by transepidermal elimination of altered collagen and/or elastin through the epidermis. These include four disease entity: Kyrle’s disease, perforating folliculitis, EPS and RPC. RPC is a rare type of perforating disorder which was first described by Mehregan et al. in 1967 [6] The underlying defect is most likely to be a genetic abnormality of the collagen leading to its extrusion as a result of necrolysis of the overlying epidermis. RPC exists in two forms; the common acquired form occuring in adults and rare inherited form seen in children. The acquired form is more common and usually manifest after second decade of life. It is often referred to as acquired perforating dermatosis mostly associated with diabetes mellitus, chronic renal failure, hypothyroidism, hyperparathyroidism, Hodgkin’s disease, neurodermatitis, IgA nephropathy, periampullary carcinoma with jaundice, hepatocellular carcinoma and HIV [7]. The inherited form is rare, usually starts in infancy or early childhood and has a slight male preponderance. The mode of inheritance may be autosomal recessive or dominant with a positive family history in about two third cases [4,8].
The clinical manifestation is similar in both acquired and inherited forms except pruritus which is often more troublesome in acquired RPC. Morphologically, lesions in both forms consist of erythematous or more commonly hyperpigmented umbilicated papules and nodules with a central keratotic plug distributed over extensor surfaces of upper and lower extremities [9]. The lesions may regress spontaneously within 2-3 months leaving a hypopigmented or hyperpigmented area [10]. In inherited and acquired RPC, physical trauma in the form of scratching and cold has been suggested as predisposing factors in inducing the degeneration of collagen with thinning of epidermis [4].
In genetically predisposed individuals in inherited RPC, these predisposing factors eventually result in early initiation of collagen damage and degeneration. [9] The transepidermal elimination of damaged collagen is facilitated by the upward movement of proliferating keratinocytes which form a vertically oriented channel through which elimination take place [9]. Overexpression of TGF-ß3 and extracellular matrix proteins like fibronectin have been held responsible to stimulate such increased keratinocytic proliferation and upward migration resulting in the perforation through epidermis [11].
Histopathological features of classical fully evolved lesions of RPC are very distinctive which shows a cup-shaped invagination of the epidermis, which is lined by acanthotic epidermis on the sides. At the base of the invagination, there are vertically oriented perforating bundles of degenerated collagen intermingled with the attenuated layer of keratinocytes [8,12].
Within the invagination, there are densely packed basophilic debris and degenerated collagen bundles. Beneath the base of the invagination, dense infiltration of lymphocytes and neutrophils may be found [12].
There are few clinical conditions which may pose some difficulty in the clinical diagnosis of RPC, they are: Kyrle’s disease, EPS, Perforating folliculitis, perforating granuloma annulare (GA), molluscum contagiosum and papular urticaria [10]. The latter three can be ruled out on the basis of characteristic clinical presentation but to differentiate from first three perforating dermatosis, histopathological evaluation is usually required. Kyrle’s disease can easily be ruled out on HPE by the absence of dyskeratotic keratinocytes & dermal granulomatous lymphocytic infiltration and presence of altered collagen bundles at the base of invagination [13].
In Perforating folliculitis, the epidermal invagination is seen in relation to a vellus hair unlike in RPC. Elastosis perforans serpiginosa is characterized by the presence of thickened elastic fibres around epidermal invaginations, which can be confirmed on Verhoeff-van Geison staining unlike in RPC [13]. Hence histopathological diagnosis may be aided by specific staining for collagen or elastin by Masson’s trichrome and Verhoeff-van Geison stain respectively.
Many treatment modalities have been tried with variable success rates. Fortunately, lesions can regress over the time without any treatment but inherited form may not resolve spontaneously so long term therapy is usually needed. The treatment options include topical retinoids, topical corticosteroids, intralesional steroids, PUVA, NBUVB, cryotherapy, allopurinol, doxycycline, methotrexate [14,15].
CONCLUSION
Inherited RPC is a rare disorder which may not resolve spontaneously leading to the persistence of the lesions and moreover it may progress over the time resulting in significant cosmetic problems with or without pruritus. Therefore in the view of such challenges with this disorder, proper case reporting should be done to allow further research and therapeutic trials to find out better treatment options.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Rapini RP, Herbert AA, Drucker CR. Acquired perforating dermatosis. Evidence for combined transepidermal elimination of both collagen and elastic fiers. Arch Dermatol. 1989;125:1074-8.
2. Faver IR, Daoud MS, Su WP. Acquired reactive perforating collagenosis. Report of six cases and review of the literature. J Am Acad Dermatol. 1994;30:575-80.
3. Bhat YJ, Manzoor S, Qayoom S, Wani R, Baba AN, Bhat AH. Familial reactive perforating collagenosis. Indian J Dermatol. 2009;54:334-7.
4. Kanan MW. Familial reactive perforating collagenosis and intolerance to cold. Br J Dermatol. 1974;91:405-14.
5. Ramesh V, Sood N, Kubba A, Singh B, Makkar R. Familial reactive perforating collagenosis: A clinical, histopathological study of 10 cases. J Eur Acad Dermatol Venereol. 2007;21:766-70.
6. Mehregan AH, Schwartz OD, Livingood CS. Reactive perforating collagenosis. Arch Dermatol. 1967;96:277-82.
7. Cerio R, Calnan CD, Wilson-Jones E. A clinico pathological study of reactive perforating collagenosis: Report of 10 cases. Br J Dermatol. 1987;117:16-7.
8. Naik NS, Nousari CH, Heilman ER, Friedman RJ. Degenerative diseases and perforating disorders. In: Elder DE, Elenitsas R, Johnson BL, Murphy GF, Xu X, editors. Lever’s Histopathology of the Skin. 10th ed. Philadelphia: Lippincott Williams and Wilkins; 2009. p. 395-6.
9. Verma R, Vasudevan B, Pragasam V, Deb P, Venugopal R, Mitra D. A rare case of familial reactive perforating collagenosis. Indian J Dermatol. 2013;58:408.
10. Pai VV, Naveen KN, Athanikar SB, Shastri DU, Rai V. Familial reactive perforating collagenosis: A report of two cases. Indian J Dermatol. 2014;59:287-9.
11. Gambichler T, Birkner L, St?ker M, Othlinghaus N, Altmeyer P, Kreuter A. Up-regulation of transforming growth factor-beta 3 and extracellular matrix proteins in acquired reactive perforating collagenosis. J Am Acad Dermatol. 2009;60:463-9.
12. Millard PR, Young E, Harrison DE, Wojnarowska F. Reactive perforating collagenosis: Light, ultrastructural and immunohistological studies. Histopathology. 1986;10:1047-56.
13. Verma R, Vasudevan B, Kakkar S, Mishra P, Pragasam V, Dabbas D. Kyrle’s disease presenting in an extensive distribution along lines of Blaschko. Indian J Dermatol. 2015;60:423.
14. Cullen S. Successful treatment of reactive perforating collagenosis with tretinoin. Cutis. 1979;23:187-91.
15. Sehgal VN, Verma P, Bhattacharya SN, Sharma S. Familial reactive perforating collagenosis in a child: Response to narrow-band UVB. Pediatr Dermatol. 2013;30:762-4.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.