|
Get Citation
|
|
|
Tiwary AK, Mishra DK. Jadassohn Lewandowsky syndrome: Type 1 pachyonychia congenita. Our Dermatol Online. 2017;8(1):56-59. |
|
|
Download citation file:
|
Jadassohn Lewandowsky syndrome: Type 1 pachyonychia congenita
Anup Kumar Tiwary, Dharmendra Kumar Mishra
Department of Dermatology, Venereology and Leprosy, Rajendra Institute of Medical Sciences, Ranchi, India
Corresponding author: Dr. Anup Kumar Tiwary, E-mail: anup07tunnu07@gmail.com
Submission: 09.02.2016; Acceptance: 18.04.2015
DOI: 10.7241/ourd.20171.15
ABSTRACT
Pachyonychia congenita (PC) is a rare, usually autosomal dominant, genodermatosis characterized by tetrad of wedge shaped nail hypertrophy, focal palmoplantar keratoderma, oral leucokeratosis and follicular hyperkeratosis due to mutation in either of the three keratin genes, KRT6, KRT16 and KRT17. Classically, it has been subdivided into 2 major types: PC-1 (Jadassohn Lewandowsky syndrome) and PC-2 (Jackson-Lawler syndrome) but, genotypically, now PC has been classified into 5 types depending upon the underlying keratin gene mutations: PC-K6a, PC-K6b, PC-K6c, PC-K16 and PC-K17. Since 1904 when Muller documented the first case of PC, around 700 cases have been reported till now. Hence, in the view of rarity of such crippling and debilitating dermatosis of congenital or early life onset, we herein report a clinically diagnosed case of Pachyonychia congenita- type 1(Jadassohn Lewandowsky syndrome) in a 16 years old girl with affection of 2 other members of her family with the same disorder.
Key words: Jadassohn Lewandowsky syndrome; Nail hypertrophy; Pachyonychia congenita; Palmoplantar keratoderma
INTRODUCTION
PC is commonly described as a rare genodermatosis characteristically manifesting as massive subungual hyperkeratosis with nail thickening, focal palmoplantar keratoderma alongwith deep fissuring and blistering, oral leukokeratosis and discrete follicular hyperkeratosis [1,2]. Other features including abnormalities of teeth, hairs and larynx can also be seen depending on the clinical types. Onset is usually within first year of life and mode of inheritance is autosomal dominant. After first description by Muller in 1904, next reports were published by Wilson in 1905 and Jadassohn Lewandowsky in 1906 [3–5]. Further, depending on the genetic mutation and clinical correlations, 4 types of PC have been defined.
CASE REPORT
A 16 yrs old girl came to our out patient department of dermatology presenting with chief complaint of hypertrophy of all the finger and toe nails with upward growth and brownish discolouration, palmoplantar keratoderma with painful blistering since the age of 1 year. The patient was born of non-consanguineous parents and normal at birth but after 1 year of age, when she started walking, recurrent blistering on soles occurred. There was no history of natal teeth. Gradually, thickening of the palm and soles developed and upward growth of nails were noticed. Then, spiny follicular hyperkeratotic lesions appeared on trunk and extremities. She also complained of intolerance to hot and spicy food. Her father and younger brother had similar affection of same degree since their infancy. She was admitted and examined thoroughly.
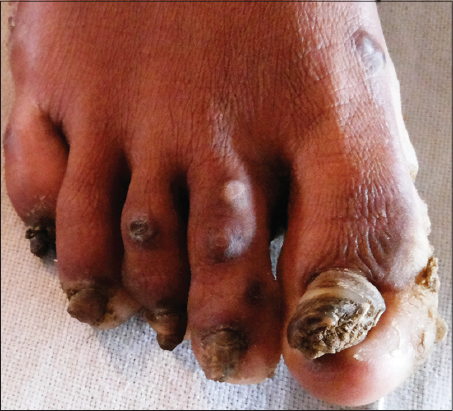
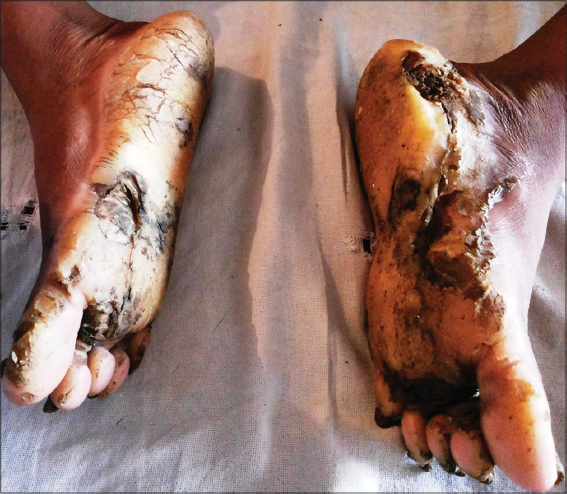
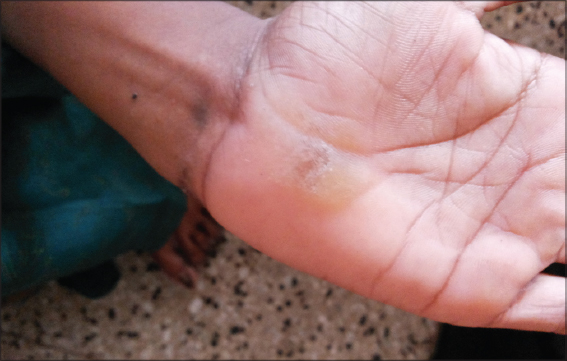
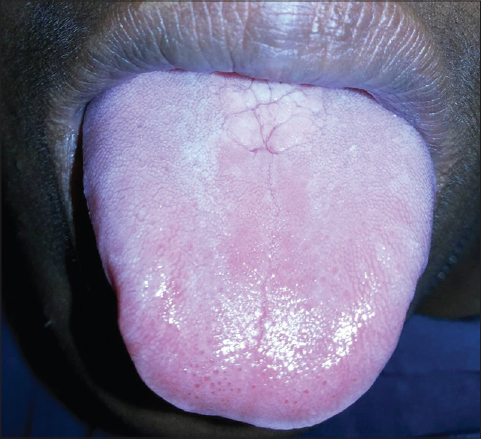
On cutaneous examination, all the fingernails and toe nails were seen to be thick, lusterless, brownish and hypertrophied and vertically grown giving a ‘door wedge shape’ to the free of the nail plate due to massive subungual hyperkeratosis (Fig. 1) Both soles were hyperkeratotic, fissured and macerated with hyperhidrosis (Fig. 2). There was minor involvement of left palm with only focal thickening (Fig. 3). The oral mucosa and tongue showed leukokeratosis with erosion (Figs. 4 and 5). She also had multiple, follicular, hyperkeratotic and spiny discrete papules over buttocks, knee and extensor aspect of thighs (Fig. 6).

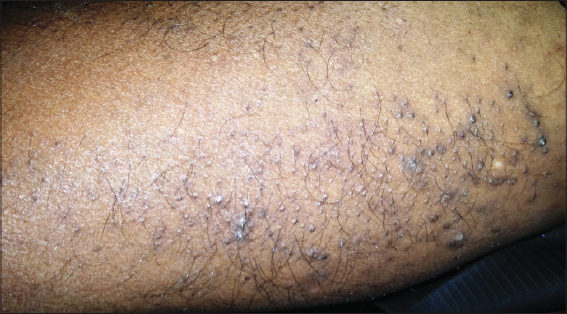
Examination of hair, teeth, throat and eyes were normal and systemic examinations were also not remarkable. There was no sign of mental retardation and all routine laboratory parameters were within normal limits. The possibility of fungal cause of nail dystrophy was ruled out by KOH microscopy.
On the basis of these clinical features, she was diagnosed with PC type 1 and started on oral isotretinoin and topical keratolytics but there was no relief and patient did not follow up.
DISCUSSION
Pachyonychia congenita is a rare group of inherited, ectodermal dysplastic disorders characterized by tetrad of wedge shaped nail hypertrophy and subungual hyperkeratosis, varying degree of focal palmoplantar keratoderma with history of recurrent blisters, oral leucokeratosis and discrete follicular papules on the sites of friction [2,3]. Other features which may be associated include alopecia, keratosis pilaris, hyperhidrosis of the palms and soles, natal teeth, laryngeal involvement causing hoarseness of voice and corneal opacities [4]. Although mode of inheritance is autosomal dominant but autosomal recessive transmission has also been seen and even sporadic cases can also occur without any family history due to spontaneous mutation. Based on the certain genetic mutations and their correlation to the presenting features,4 clinical types or variants have been proposed over the time [5].
- PC tupe-1 (Jadassohn-Lewandowski syndrome) – is a relatively common type, characterized by upward growth of thick, friable and lusterless discoloured nails of all the digits (most severely of thumb and index finger), palmoplantar keratoderma with hyperhidrosis in more than half of the cases, follicular hyperkeratosis, oral or laryngeal leukokeratosis and blisters [6].
- PC type-2 (Murray–Jackson– Lawler syndrome) – natal teeth and cysts like epidermoid cysts, eruptive vellus hair cyst or steatocytoma multiplex along with less prominent features of PC type 1 [2].
- PC type-3 (Schafer–Brunauer syndrome) – characterized by features of types 1 and 2 with angular chielitis, corneal dyskeratosis and cataracts [1].
- PC type-4 (PC tarda) – This type was suggested by Paller et al. in 1991, having late onset (during late childhood or adulthood) and manifest features of all three types with laryngeal involvement, mental retardation and hair changes like sparse kinky hairs and alopecia [7,8].
- PC with only nail involvement has also been reported.
Aetiopathologically, around 100 mutations have been identified which can lead to the development of such deformities. The keratin genes which are expressed on palm, soles, nail bed, oral mucosa and pilosebaceous units and responsible for this syndrome are- KRT6a, KRT6b, KRT6c, KRT16 and KRT17 [5,6]. These mutations interferes with the assembly of polypeptides forming the keratin skeleton of epidermal cells. Histological examination of keratoderma is non-specific with the demonstration of orthokeratosis and parakeratosis, non epidermolytic acanthosis [9]. Cytological atypia is usually not seen but the possibility of malignant changes in palmoplantar lesions can not be neglected over the time. Although PC is usually diagnosed on the clinical grounds, only genetic testing can confirm the diagnosis and types of PC due to the overlapping clinical features.
Treatment modalities are chiefly meant for hyperkeratosis and includes topical keratolytics (salicylic acid, urea, lactate), oral retinoic acid derivatives (isotretinoin, acitretin), mechanical abrasion with hand tool or by dermabrasion, electrofulguration and excision. Treatment of aggravating factors like hyperhidrosis by means of aluminium chloride lotion, iontophoresis or botulinum toxin injection may provide pain-relief and also reduce blistering.
Research work on gene therapies are being carried out to make ‘inhibition of mutant allele’ possible.
CONCLUSION
Being a very rare, genetic and crippling condition and having only old, conventional and very few treatment options, every clinically diagnosed case of PC should be reported and efforts should be made for genetic testing so as to help advancement in the genetic therapies.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. McLean WH, Hansen CD, Eliason MJ, Smith FJ, The phenotypic and molecular genetic features of pachyonychia congenitaJ Invest Dermatol 2011; 131: 10157-
2. Leachman SA, Kaspar RL, Fleckman P, Florell SR, Smith FJ, McLean WH, Clinical and pathological features of pachyonychia congenitaJ Investig Dermatol Symp Proc 2005; 10: 317-
3. Fernandez RJ, Parikh DA, Pachyonychia CongenitaIndian J Dermatol Venereol Leprol 1989; 55: 334-5.
4. Rodríguez H, Cuestas G, Zanetta A, Balbarrey Z, Pachyonychia congenita with involvement of the larynxActa Otorrinolaringol Esp 2013; 6519: 2646-
5. Munro CS, Pachyonychia congenita:Mutations and clinical presentationsBr J Dermatol 2001; 144: 929-30.
6. Al Aboud A, Al Aboud K, Josef Jadassohn (1?936), Felix Lewandowsky (18791921), and their syndromeClin Cosmet Investig Dermatol 2011; 4: 17982-
7. Vaccaro M, Guarneri F, Barbuzza O, Guarneri C, Pachyonychia congenita tarda affecting only the nailsDermatol Online J 2008; 14: 12-
8. Moger G, Shashikanth MC, Chandrashekar KT, Kurein S, Pachyonychia congenita tarda:A rare case reportContemp Clin Dent 2013; 4: 409-11.
9. Johnson BL, JrYan AC, Elder DE, Congenital diseases (Genodermatosis)Lever’s Histopathology of the Skin 2009; 10th ed. Philadelphia: Lippincott Williams and Wilkins; 138-
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.