|
Get Citation
|
|
|
Tiwary AK, Mishra DK, Chaudhary SS. Familial dyskeratotic comedones in a female with positive family history: A rare entity. Our Dermatol Online. 2017;8(1):46-48. |
|
|
Download citation file:
|
Familial dyskeratotic comedones in a female with positive family history: A rare entity
Anup Kumar Tiwary, Dharmendra Kumar Mishra, Shyam Sundar Chaudhary
Department of Dermatology, Venereology and Leprosy, Rajendra Institute of Medical Sciences, Ranchi, India
Corresponding author: Dr. Anup Kumar Tiwary, E-mail: anup07tunnu07@gmail.com
Submission: 04.03.2016; Acceptance: 06.06.2016
DOI: 10.7241/ourd.20171.12
ABSTRACT
Familial dyskeratotic comedones is an inherited disorder with characteristic clinical features characterized by disseminated, hyperkeratotic papules and comedones with evidence of dyskeratosis on histopathology. In the light of unrewarding treatment and rarity of this entity, herein we report this rare disorder in a female patient having positive family history.
Key words: Comedones, dyskeratotic, hyperkeratotic papules
INTRODUCTION
Rodin et al. first reported a rare inherited condition in 1967, called as familial dyskeratotic comedones (FDC) with autosomal dominant mode of inheritance and clinically characterized by symmetrical development of cosmetically disfiguring, progressive but asymptomatic, numerous, discrete, disseminated, hyperkeratotic papules and comedones on trunk, arms and face, appearing around puberty [1]. Histopathological examination often reveal craterlike epidermal invaginations plugged with lamellar keratinous materials and dyskeratotic cells. To the best of our knowledge, less than 50 cases of familial dyskeratotic comedones are reported in literature. Here we report a female case with this rare disorder having a positive family history.
CASE REPORT
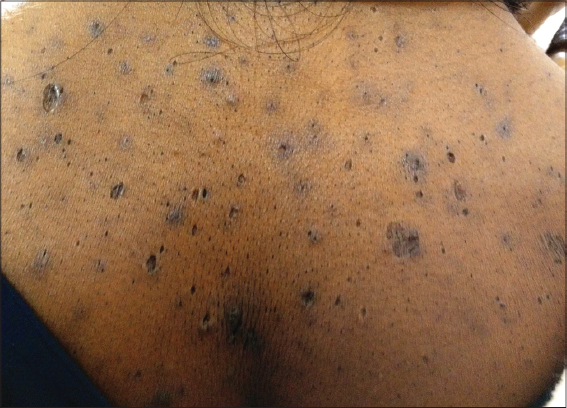
A 17 year old female presented with multiple, asymptomatic hyperkeratotic and hyperpigmented comedone-like papules and atrophic pock-like scars predominantly over central face, neck, chest, abdomen, upper extremities. These lesions started appearing on face, trunk and then upper limbs since the age of 11 years which gradually increased in number and severity with some exacerbations and remissions and forming multiple pock-like scars chiefly over face and back with no evidence of acne or other relevant dermatoses. There was a family history of similar lesions in her older sister and maternal grandmother since their childhood. There was no history of consanguinity among her parents.
Cutaneous examination showed numerous, hyperkeratotic papules, comedones and pock-like scars of size ranging from 0.5 to 2 cm on central area of face, neck, chest, abdomen, upper extremities and buttocks sparing scalp, legs, palms, soles, genitalia and mucosa. (Figs. 1 and 2). Systemic examination and routine laboratory parameters were within normal limits.

To reach at a final diagnosis, a punch biopsy was done and two samples were taken from comedone-like lesion and scarred lesion and sent for histopathological examination.
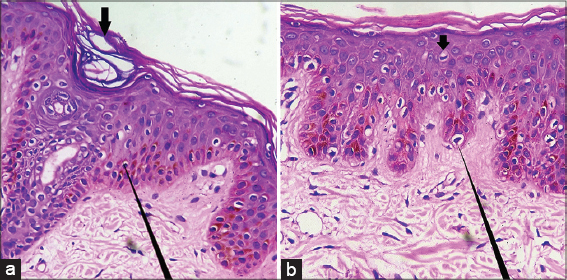
Histopathological evaluation demonstrated hyperkeratosis, multiple crater-like epidermal invaginations containing plugs of lamellar keratinous materials. Acantholysis was absent and dyskeratosis was observed at some places (Figs. 3a and 3b).
DISCUSSION
Although first reported in 1967, Carneiro et al. was the first who described this rare entity in a family of four affected members and proposed the term ‘familial dyskeratotic comedones’ in 1972 based on the following distinctive clinical features [2]:
- Lesions clinically resembling comedones
- Occurrence in some family members
- Presence of dyskeratotic changes on histological examination.
In 1999, McKusick kockaert proposed this disease to be inherited in autosomal dominant mode of inheritance so complete family history should always be taken [3]. Clinically, the lesions usually appear around puberty and chiefly involve face, trunk and upper extremities in progressive fashion [3]. The classical lesions of acne is usually not found to be present [4–6]. This condition is mostly asymptomatic but occasionally may be associated with pruritus, pain and burning sensation due to the inflammation [3,4,7]. Histopathologically it is characterized by hyperkeratosis, multiple crater-like epidermal invaginations containing plugs of lamellar keratinous materials, acantholysis and dyskeratosis [4,7]. On electron microscopy, FDC shows a reduced number of desmosomal attachments within the stratum malpighii leading to suprabasal acantholysis but not in all cases as in our case [4].
There are many closely simulating clinical conditions which should be kept in differential diagnoses. They are acne vulgaris, naevus comedonicus, keratosis pilaris (KP), Darier’s disease, kyrle’s disease, reactive perforating collagenosis (RPC), perforating folliculitis and recently added entity in the literature ‘familial disseminated comedones without dyskeratosis’ [1,2,5,6–8]. Comedonal Darier’s disease presents with follicular and extrafollicular greasy, hyperkeratotic papules and plaques in seborrheic areas shows dyskeratotic cells like corps ronds and grains, suprabasal acantholysis and villi, which are diagnostic [9]. Nevus comedonicus has early life onset and presents with closely arranged, dilated follicular openings with keratinous plugs predominantly over face and neck; mostly unilateral distribution. Kyrles disease, RPC and KP can be easily differentiated on histopathology.
Hence, on the basis of peculiar clinical features, mode of inheritance and distinctive histopathological findings, diagnosis of FDC was made in our case.
Treatment is a tough task because none of the available treatment modalities are effective. Treatment with topical and oral retinoic acid derivatives are also unrewarding because of the different pathophysiological process in FDC from that of normal comedones in acne [4,5–7]. However, frequent sun exposure and carbon dioxide laser have shown good results [4,7].
CONCLUSION
Being asymptomatic and having good prognosis, this rare entity may easily be overlooked and under reported. But in view of social problems especially in the female patients due to the severe involvement of the face leading to the psychological distress and disability of pateints, further studies are much needed to accumulate more knowledge about the pathogenesis of this disease to find out the definitive cure.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Rodin HH, Blakenship ML, Bernstein G, Diffuse familial comedonesArch Dermatol 1967; 96: 1456-
2. Carneiro SJ, Dickson JE, Knox JM, Familial dyskeratotic comedonesArch Dermatol 1972; 105: 24951-
3. Van Geel NA, Kockaert M, Neumann HA, Familial dyskeratotic comedonesBr J Dermatol 1999; 140: 9569-
4. Hall JR, Holder W, Knox JM, Verani R, Familial dyskeratotic comedones. A report of three cases and review of the literatureJ Am Acad Dermatol 1987; 17: 80814-
5. Kumaran MS, Appachu D, Jayaseelan E, Familial dyskeratotic comedonesIndian J Dermatol Venereol Leprol 2008; 74: 1424-
6. Cheng MJ, Chen WC, Happle R, Song ZQ, Familial disseminated comedones without dyskeratosis:Report of an affected family and review of the literatureDermatology 2014; 228: 3036-
7. Hallermann C, Bertsch HP, Two sisters with familial dyskeratotic comedonesEur J Dermatol 2004; 14: 2145-
8. Cantu DJ, Gomez-Bustamente MO, Gonzalez-Mendoza A, Snchez-Corona J, Familial comedonesArch Dermatol 1978; 114: 1807-9.
9. Chung J, Kim JY, Gye J, Namkoong S, Hong SP, Park BC, Acase of familial comedonal Darier’s diseaseAnn Dermatol 2011; 23: Suppl 3S398401-
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.