|
Get Citation
|
|
|
Funda Tamer, Mehmet Eren Yuksel. A case of multiple lipomas coexisting with angiolipomas: Familial multiple lipomatosis or a distinct entity?. Our Dermatol Online. 2016;7(4):439-441. |
|
|
Download citation file:
|
A case of multiple lipomas coexisting with angiolipomas: Familial multiple lipomatosis or a distinct entity?
Funda Tamer1, Mehmet Eren Yuksel2
1Department of Dermatology, Turgut Ozal University Faculty of Medicine, Alparslan Turkes Caddesi, No: 57 Emek, Ankara, Turkey, 2Department of General Surgery, Devrek State Hospital, Devrek, Zonguldak, Turkey
ABSTRACT
Familial multiple lipomatosis and familial multiple angiolipomatosis are both rare and autosomal-dominantly inherited diseases. Familial multiple lipomatosis is characterized by multiple lipomas on the trunk and extremities, while familial multiple angiolipomatosis presents with multiple angiolipomas. Hereby, we present a 50-year-old Caucasian male patient with both multiple lipomas and angiolipomas on his trunk, arms and thighs with a family history of similar subcutaneous lesions. As multiple lipomas and angiolipomas coexist in the same patient, we suggest that this condition should be nominated as a distinct entity rather than a traditional familial multiple lipomatosis case.
Key words: Familial; Multiple; Lipomatosis; Angiolipomatosis
INTRODUCTION
Familial multiple lipomatosis is a rare, autosomal dominant inherited disease with an incidence of 0.002% in the general population. It is characterized by multiple, mobile, encapsulated subcutaneous nodules on forearms, thighs and trunk. On the other hand, familial multiple angiolipomatosis is an autosomal dominant inherited disease presenting with several angiolipomas typically on arms and trunk. A lipoma with an excessive vascular component is called an angiolipoma. Therefore, familial angiolipomatosis has been usually considered as subtype of familial multiple lipomatosis.
However, lipomas and angiolipomas may coexist. Hereby, we present a 50-year-old Caucasian male patient suffering from both multiple lipomas and angiolipomas on his trunk, arms and thighs with a family history of subcutaneous nodules. As multiple lipomas and angiolipomas coexist in the same patient, we suggest that this condition should be described as a distinct entity rather than a traditional familial multiple lipomatosis case.
CASE REPORT
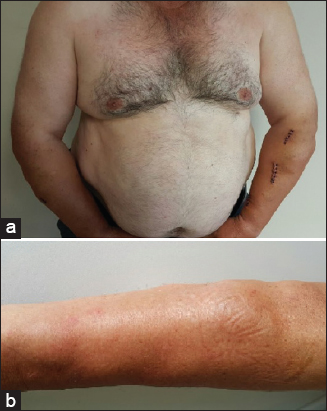
A 50-year-old Caucasian male patient complaining of multiple swellings on his trunk, arms and legs which had been slowly increased in size and number in the last 35 years, was admitted for further clinical evaluation. The patient stated that the lesions were painless, however he had cosmetic concerns. The past medical history was unremarkable. The patient admitted that his two sisters had similar swellings on their arms and legs. The physical examination revealed multiple, skin colored, mobile, soft, well defined, round shaped subcutaneous nodules (Figs. 1a and 1b). Laboratory tests including total cholesterol and LDL cholesterol levels were within normal limits, 177 mg/dl and 96 mg/dl respectively (reference ranges of total cholesterol and LDL cholesterol are 10-200 mg/dl and 10-150 mg/dl respectively). However, serum level of triglyceride was 279 mg/dl (normal range is 10 to 150 mg/dl) which was higher than the normal range. HDL cholesterol was 25 mg/dl (normal range is 30 to 80 mg/dl) which was lower than the normal range. In addition, serum level of hemoglobin was 17 g/dL (normal range is 10 to 16,5 g/dL) and aspartate transaminase was 83 u/l (normal range is 5 to 34 u/l). As triglyceride and aspartate transaminase levels were higher than expected, hepatosteatosis was initially suspected. Therefore, an abdominal ultrasonography was performed. It revealed multiple abdominal echogenic subcutaneous nodules of 2.5 cm maximum diameter, hepatomegaly and grade 2 hepatosteatosis.
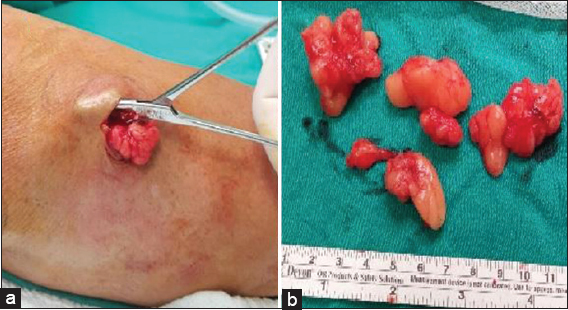
As the patient had cosmetic concerns, he demanded the surgical removal of the lesions. Therefore, 18 yellowish brown colored nodules on the patient’s arms with a minimum of 0,8×0,4×0,2 cm and a maximum of 4,5x3x1,5 cm in size were surgically removed under local anesthesia (Figs. 2a and 2b). The patient did well postoperatively. The histopathological examination of 10 of 18 lesions revealed lipomas and the 8 of 18 lesions were found to be angiolipomas.
DISCUSSION
Familial multiple lipomatosis is a rare hereditary disorder which is characterized by multiple lipomas mainly on trunk and extremities. It has been first reported by Brodie as “lipomatose circonscrite multiple” in 1846 [1]. It is a matter of debate whether familial multiple lipomatosis is associated with abnormalities of lipid metabolism. Some of the authors like Wilson and Rubinstein mentioned that familial multiple lipomatosis might be associated with hyperlipidemia [2,3]. However, the patient presented above had hypertriglyceridemia and low HDL cholesterol levels.
Moreover, familial multiple lipomatosis might coexist with other diseases. Celiac disease and cytophagic histiocytic panniculitis have been reported in patients with familial multiple lipomatosis [4,5]. Recently, Nunes et al. described a female patient who had brain lipoma, corpus callosum hypoplasia, polymicrogyria and familial multiple lipomatosis [6]. In addition, Madelung’s disease, Dercum’s disease, Bannayan-Zonana syndrome and Cowden syndrome clinically present with subcutaneous lipomas. Therefore, they should be kept in mind in the differential diagnosis of familial multiple lipomatosis. Surgical excision, liposuction and lipolysis with phosphatidyl choline injection are some of the options in the treatment of familial multiple lipomatosis [7].
On the other hand, familial multiple angiolipomatosis is a benign, autosomal dominant condition which clinically presents with numerous angiolipomas. Angiolipoma is a rare variant of lipoma with excessive vascular component. Angiolipomas can cause pain and thus, it may be helpful to differentiate angiolipomas from lipomas clinically. Besides, they usually do not require treatment like lipomas. Therefore, familial angiolipomatosis is generally considered as a subtype of familial multiple lipomatosis [7,8]. However, Hapnes et al. and Kumar et al. regard familial multiple angiolipomatosis as a distinct disease [9,10].
The case presented above is unique as the patient had both multiple lipomas and angiolipomas with a family history of subcutaneous nodules. In addition to familial lipomatosis and familial angiolipomatosis, we suggest that the coexistence of lipomas and angiolipomas should be classified as a distinct entity.
CONSENT
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Gologorsky Y, Gologorsky D, Yarygina AS, Surti U, Zirwas MJ, Familial multiple lipomatosis: Report of a new familyCutis 2007; 79: 227-32.
2. Wilson D, Boland J, Sporadic multiple lipomatosis: a case report and review of the literatureW V Med J 1994; 90: 145-6.
3. Rubinstein A, Goor Y, Gazit E, Cabili S, Non-symmetric subcutaneous lipomatosis associated with familial combined hyperlipidaemiaBr J Dermatol 1989; 120: 689-94.
4. Sayar I, Demirtas L, Gurbuzel M, Isik A, Peker K, Gulhan B, Familial multiple lipomas coexisting with celiac disease: A case reportJ Med Case Rep 2014; 8: 309-
5. Krilis M, Miyakis S, Cytophagic histiocytic panniculitis with haemophagocytosis in a patient with familial multiple lipomatosis and review of the literatureMod Rheumatol 2012; 22: 158-62.
6. Nunes JC, Martins RF, Bastos A, Claudino LS, Guarnieri R, Lima Dde C, Brain lipoma, corpus callosum hypoplasia and polymicrogyria in familial multiple lipomatosisClin Neurol Neurosurg 2013; 115: 1157-9.
7. Rosmaninho A, Pinto-Almeida T, Fernandes IC, Machado S, Selores M, Do you know this syndrome?An Bras Dermatol 2012; 87: 324-5.
8. Abbasi NR, Brownell I, Fangman W, Familial multiple angiolipomatosisDermatol Online J 2007; 13: 1-3.
9. Hapnes SA, Boman H, Skeie SO, Familial angiolipomatosisClin Genet 1980; 17: 202-8.
10. Kumar R, Pereira BJ, Sakhuja V, Chugh KS, Autosomal dominant inheritance in familial angiolipomatosisClin Genet 1989; 35: 202-4.
Notes
Source of Support: Nil
Conflict of Interest: None declared.
Comments are closed.