Primary idiopathic systemic amyloidosis – Rare classical cases with fatal outcome
Shivakumar Patil, Supraja Chinthala, Snehal Lunge
Department of Dermatology, Kle University, Jawaharlal Nehru Medical College, Belgaum, India
ABSTRACT
Primary systemic amyloidosis is a rare condition. We report two cases of primary systemic amyloidosis. Both the cases were without any hematological abnormality. Cutaneous features were the predominant presenting symptoms in these patients. The patients presented with typical waxy lesions on face and macroglossia. Diagnosis was confirmed by histopathology with haematoxylin and eosin staining and Congo red staining.
Key words: Primary systemic amyloidosis; Macroglossia; Congo red stain
INTRODUCTION
Amyloidosis is a generic term originally coined by Rudolf Virchow in 1854, which denotes extracellular deposition of a proteinaceous substance composed of one of a family of biochemically unrelated proteins depending on the underlying condition, and which is associated with considerable tissue dysfunction [1]. Amyloid deposition may be localized to one organ system or it may involve multiple organs depending on which amyloidosis is classified as localized or systemic. Localized cutaneous amyloidosis, which includes macular amyloidosis and lichen amyloidosis, is a benign common disease without systemic involvement. Skin may also be involved in systemic amyloidosis. Systemic amyloidosis is classified into primary, secondary and familial [2]. Primary systemic amyloidosis may be idiopathic or myeloma-associated. Secondary systemic amyloidosis is associated with many chronic inflammatory disorders. Skin involvement is common in primary amyloidosis whereas secondary amyloidosis rarely involve skin [1]. Systemic amyloidosis is usually a disease of the elderly.
CASE REPORTS
Case 1
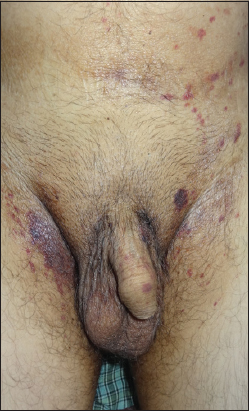
The first case of 61 year old male presented with features of asymptomatic waxy papules on the face especially involving the periorbital areas (Fig. 1) and features of macroglossia (Fig. 2) were seen along with purpuric lesions (Fig. 3) involving the face, neck and groin. Thickened palms and fingers associated with burning sensation, difficulty in moving fingers.
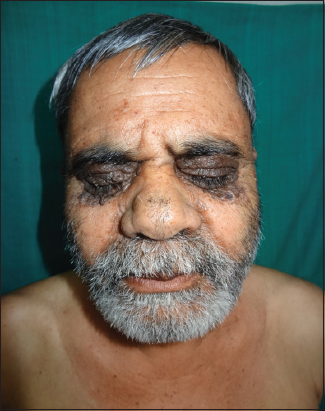

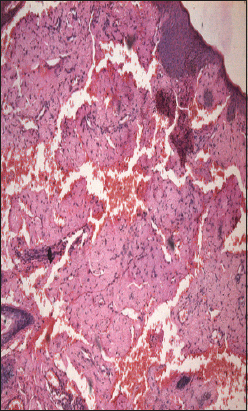
Other changes seen were Hepatomegaly, left ventricular hypertrophy and 10% plasmacytoma was seen on bone marrow biopsy. Evidence of renal failure like raised blood urea and serum creatinine along with urine albumin was present. Because of the typical presentations, diagnosis of primary systemic amyloidosis, was suspected clinically and confirmed by histopathology. Histopathological features noted were epidermis showing hyperkeratosis and focal atrophy; dermis showing diffuse deposits of eosinophilic hyaline material extending into subcutaneous tissue (Fig. 4).
Case 2
36 year old male presented with Weight loss, weakness, fatigue, and asymptomatic raised lesions on face since 3 months.
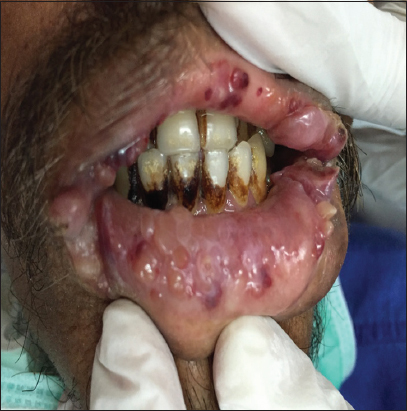
On examination Macroglossia with lateral teeth indentations and fissures on tongue (Fig. 5).
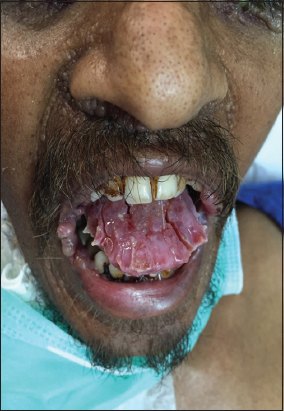
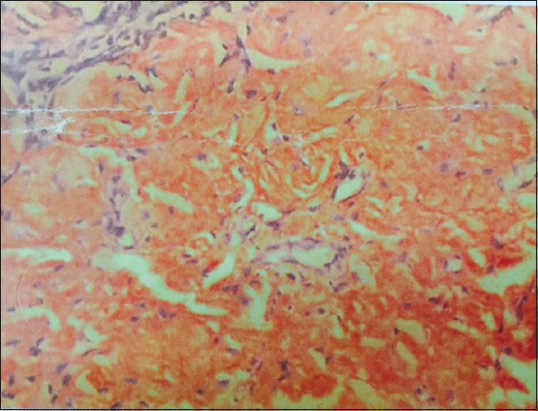
Multiple waxy papules, nodules and tumefactive lesions in oral mucosa (Fig. 6), periorbital and perioral areas (Fig. 7). Ecchymotic patches were seen on face and trunk. Investigations revealed a low complete blood count with Haemoglobin of 7 gm/dl. Blood urea of 89 mg/dl, Serum creatinine. 2.8 mg/dl and Urine albumin – 3+, serum and urine electrophoresis was normal. USG abdomen revealed grade 1 renal parenchymal disease. Biopsy H and E staining – Eosinophilic amorphous material. Congo red staining – brick red staining of amyloid (Fig. 8). Bone marrow examination was normal.

Prior to the study, patient gave written consent to the examination and biopsy after having been informed about the procedure.
DISCUSSION
Amyloidosis is a group of diseases characterized by extracellular deposition of beta-sheet fibrils. In the systemic forms, the amyloid causes progressive organ dysfunction leading to death of the patients. Over 20 proteins capable of amyloid formation have been identified. They include immunoglobulin (Ig) light chains in primary systemic amyloidosis (AL), Ig heavy chain (AH), amyloid A in secondary amyloidosis (AA), beta2-microglobulin in dialysis-associated arthropathy (Ab2M) and amyloid beta protein (Ab) in Alzheimer’s disease and Down’s syndrome. There are also hereditary forms that include transthyretin (ATTR), apoli-poprotein A-I (AApoAI) and A-II (AApoAII), gelsolin (AGel), lysozyme (ALys), fibrinogen A-alfa chain (AFib) and others. Another amyloidogenic protein is leukocyte chemotactic factor 2 (LECT2) [3].
Inherited amyloidosis is due to mutation in certain precursor protein, which makes them susceptible to mis- folding. In case of primary systemic amyloidosis, the amyloid is derived from monoclonal immunoglobulin light chain and is called as AL amyloid where L stands for light chain of immunoglobulin molecule. In case of secondary amyloidosis which is associated with many chronic inflammatory diseases, amyloid fibrils are derived from cleavage fragment of the circulating acute phase reactant serum amyloid A protein (SAA), hence called as AA amyloid. Serum amyloid A protein is synthesized in liver during inflammation [2]. It has been proposed that in macular and lichen amyloidosis, focal epidermal damage and filamentous degeneration of keratinocytes is followed by apoptosis and conversion of filamentous masses (colloid bodies) into amyloid material in the papillary dermis [1]. The reason that many diverse conditions are associated with amyloidosis may be because each of these conditions results in excessive production of proteins that are prone to mis-folding [2] In multiple myeloma-associated AL amyloidosis, precursor light chains of immunoglobulin (Bence Jones protein) are produced in large quantity by malignant plasma cell clone and can be detected in serum or urine by electrophoresis. Multiple myeloma is a malignancy of plasma cell. Amyloidosis develops in about 15% of patients of myelomatosis [1] Majority of patients of AL amyloidosis do not have obvious B-cell/plasma cell neoplasm (idiopathic). These patients might have underlying B-cell dyscrasia in which production of abnormal protein, rather than production of tumor masses, is the predominant manifestation [2]. In one study from Lebanon, of 39 cases of systemic amyloidosis, 21 were of AL type and out of these 21 cases of AL amyloidosis, 9 (43%) were associated with multiple myeloma and 12 (57%) were idiopathic [4].
Cutaneous involvement is seen in 40% patients with AL amyloidosis. Cutaneous manifestation depends upon the site of amyloid deposited. Amyloid deposition in superficial dermis produces shiny waxy translucent papules and common sites for predilection are eyelids, retro-auricular areas, neck, axilla. Infiltration of nail matrix by amyloid may produce ridging, splitting and brittleness of nail plate [1]. Similar lesions and distribution was noted in both our patients and nails were found to be dystrophic. Amyloid deposited around pilosebaceous unit leads to the destruction of hair, producing alopecia. Diffuse infiltration of scalp skin results in the enlargement of skin which gets thrown into longitudinal folds resembling cutis vertices gyrata. Diffuse infiltration of large area of skin may simulate scleroderma [1].
Amyloid infiltration of vessel wall causes capillary wall fragility, which leads to purpura and ecchymosis after a minor trauma or even spontaneously. Periorbital area is one of the common sites of expression of purpura. The capillary fragility may be demonstrated by pinching the skin. Ecchymotic lesions were present in both cases. Purpuric lesions with normal platelet count and normal coagulation profile should suggest the possibility of capillary fragility [1].
Amyloid deposition in tongue leads to macroglossia. Tongue is diffusely enlarged and firm and there may be tooth indentation along its lateral border. Amyloidosis is the commonest cause of macroglossia in adults [1]. Macroglossia if severe might lead to dysphagia. Macroglossia with tooth indentation was present in second case.
Hepatomegaly occurs in 50% of patients and splenomegaly in 10%. Hepatomegaly was present in our case 1. Cardiac involvement leads to conduction defects, arrhythmias, congestive cardiac failure and may account for 40% of deaths. Our first patient had left ventricular hypertrophy and second patient had no cardiac involvement as indicated by normal ECG and X-ray chest.
Carpal tunnel syndrome is seen in up to 25% of patients of primary systemic amyloidosis [1], as was present in our case 2. Renal involvement presents with proteinuria and renal failure. It is one of the bad prognostic indicator and was present in our case 2 as indicated by proteinuria and USG findings.
Both the patients were diagnosed as having primary amyloidosis on clinical ground. Diagnosis was confirmed by demonstration of amyloid in skin biopsy. Clinically, it is difficult to distinguish primary, secondary or familial form of amyloidosis. Immunohistochemical staining using commercially available antisera is useful for classifying the type of amyloid deposited in tissues [5], which was not done in our patients, this being a resource poor setting. Biopsy is very important for the diagnosis. Hematoxylin and eosin staining suggests the possibility of amyloidosis but Congo red staining confirms the diagnosis. Congo red staining results in a brick red color of amyloid when seen under ordinary light and under polarized light shows classical green birefringence. Unfortunately, polarized microscopy is not easily available in developing country like India. In systemic amyloidosis, amyloid deposits are seen in dermis, subcutaneous tissue and blood vessels, where as in localized cutaneous amyloidosis, deposits are seen only in papillary dermis; subcutaneous tissues and blood vessels are not involved.
Prognosis in AL amyloidosis is poor and major causes of death are cardiac and renal failure. The median survival of patients with myeloma-associated amyloidosis is five months and 2.1 years for patients with primary systemic amyloidosis [1]. Prognosis depends upon the extent of involvement. Treatment of amyloidosis is aimed at reducing the supply of precursor proteins [1]. In AL amyloidosis, the precursor is immunoglobulin light chain produced by B lymphocytes/plasma cells hence treatment with cytotoxic agents like melphalan and prednisolone that reduces plasma cell proliferation is useful [1]. Chemotherapy will be useful only when precursors are supplied by plasma cells like AL amyloidosis.
In localized cutaneous amyloidosis, such as lichen amyloidosis and macular amyloidosis, where precursors are derived from keratinocytes and not from plasma cells, alkylating agents or any other chemotherapeutic agents will not be beneficial and may be harmful [6].
These cases of systemic amyloidosis are presented for its rare occurrence. High index of suspicion is necessary for the diagnosis of such rare cases.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Breathnach SM, Burns T, Breathnach S, Cox N, Griffiths C, Metabolic and Nutritional DisordersRook’s Textbook of Dermatology 2004; 7th ed. Oxford: Blackwell publishing; 57.36-57.51.2010
2. Kumar V, Abbas A, Fausto N, Kumar V, Abbas A, Fausto N, Diseases of ImmunityRobbins and Cortran’s Pathologic Basis of Diseases 2005; 7th ed. Philadelphia: WB Saunders; 258-64.
3. Abdallah E, Waked E, Incidence and clinical outcome of renal amyloidosis: A retrospective studySaudi J Kidney Dis Transpl 2013; 24: 950-8.
4. Saba M, Tohmé A, Abadjian G, Haddad F, Ghayad E, Multisystemic amyloidosis. Clinical study of 39 patients in LebanonPresse Med 2005; 34: 640-6.
5. Gertz MA, The classification and typing of amyloid depositsAm J Clin Pathol 2004; 121: 787-9.
6. Anesi E, Palladini G, Perfetti V, Therapeutic advances demand accurate typing of amyloid depositsAm J Med 2001; 111: 243-4.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.