Hutchinson-Gilford progeria syndrome: a rare case report
Kalegowda Deepadarshan1, Bugude Gangadhar2, Mallaiah Mallikarjun2
1Department of Dermatology, Mandya Institute of Medical Sciences, Mandya-571401, Karnataka, India, 2Department of Dermatology, KVG Medical College and Hospital, Sullia, DK.-574327, India
ABSTRACT
Progeroid syndromes are characterised by clinical features of physiological aging at an early age. Hutchinson-Gilford progeria syndrome is a type of progeroid syndrome, characterised by abnormal facies, bone abnormalities, sclerodermatous skin changes and retarded physical development. Average life expectancy of progeria patients is 13 years. Herein we are reporting a case of progeria who is 21 years old.
Key words:Hutchinson-Gilford progeria syndrome; Short stature; Scleroderma
INTRODUCTION
Huthinson-Gilford progeria syndrome is a rare hereditary disorder caused by mutations in lamin A gene which encodes proteins of nuclear lamina, results in production of a truncated protein called progerin [1]. This disorder was first reported by Hutchinson in the year 1886 [2]. Gilford introduced the term progeria. This syndrome along with Werner syndrome are also known as segmental aging syndromes, as they do not feature all aspects associated to physiological aging. Till now less than 100 cases of progeria have been reported in world literature [3].
CASE REPORT
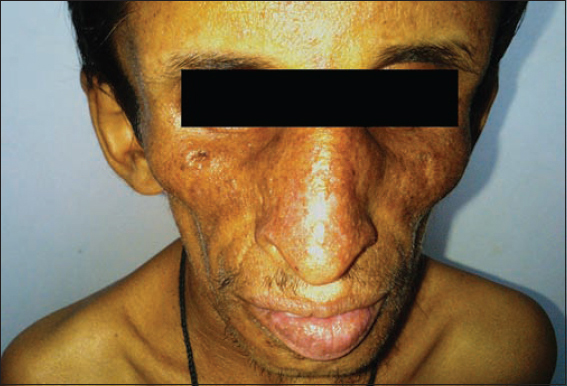
A 21 year old male presented with pigmentation of face with itching as major complaint since 6 months. No history of photosensitivity. There was history of delayed physical development and dentition noted at 2yrs of age with no history of convulsions. Intelligence was normal. On examination he had typical bird like facies characterised by large cranium with frontal bossing, prominent eyes, beaked nose, prominent ears, sunken cheeks, micrognathia and mottled pigmentation (Fig. 1). In our patient, unlike progeria there was no loss of eyebrows and scalp hair.
Detailed examination revealed short stature with height measuring 94 cm (Fig. 2). Nails were thin and brittle. Sclerodermatous thickening of skin noted over the dorsa of hands and fingers. Prominent joints, loss of subcutaneous fat and muscle atrophy were other important features noted. Ocular examination was within normal limit. Systemic examination were within normal limits. Radiological examination of skull, chest and extremities were within normal limits. Electrocardiography showed features of left ventricular failure.

DISCUSSION
Hutchinson-Gilford progeria syndrome(HGPS) is a rare disorder, has an incidence of 1 in 4 to 8 billion newborns [4]. It occurs due to sporadic mutation, hence not run in families. Characterised by accelerated aging, short stature, alopecia, generalised atrophy of skin and muscles with sclerodermatous changes over the extremities. The facial appearance is reminiscent of a fledgling bird, with a disproportionately large cranium with patent fontanelles and frontal bossing, prominent eyes and scalp veins, very sparse, downy scalp hair, sparse or absent eyebrows and eyelashes, centrofacial cyanosis, micrognathia, thin lips and a ‘beaked’ nose [5,6]. Bird like facies is not characteristic of HGPS, also seen in association with Hallermann-Streiff syndrome, familial partial lipodystrophy, Nijmegan breakage syndrome [7].
Progressive mottled hyperpigmentation of skin is noted over exposed parts with aging, but there is no photosensitivity. Abnormal and delayed dentition is noted and associated skeletal abnormalities such as dystrophic clavicles and coxa valga, with joint contractures and a ‘horse-riding’ stance also observed.
It has to be differentiated from other aging syndrome like Werner’s syndrome (adult progeria) which usually presents between 14 and 18 years of age with short stature, immature sexual development, cataract, glaucoma, and sclerodermatous changes, Cockyane’s syndrome manifested as facial erythema in butterfly distribution, photosensitivity, ocular defects, and a “micky mouse” appearance with disproportionately large hands and feet and protruding ears, Rothmund-thomson syndrome appears between 3 and 6 months of age with poikilodermatous skin, photosensitivity, and cataract.
Our patient had retarded growth and accelerated aging since 2yrs of age, with typical facial features except for presence of eyebrows and scalp hair, short stature, sclerodermatous changes in skin, loss of subcutaneous fat and atrophy of skin and muscles, radiological features of osteopenia with features of left ventricular failure in electrocardiogram.
Complications include atherosclerosis, angina attacks, cerebrovascular accidents, osteoporosis, pathological fractures and dental anomalies.
Treatment is mainly symptomatic. Long term follow up of patient is required to observe skeletal and cardiovascular changes. Main aim of treatment is to control diabetes, osteoporosis and atherosclerotic changes. Low dose aspirin is used prophylactically to control ongoing atherosclerosis. Pravastatin and bisphosphonates like zoledronic acid to prevent atherosclerosis and osteoporosis is under trial. A phase II trial of lonafarnib, a farnesyl transferase inhibitor, in progeria is currently ongoing in USA [4].
CONCLUSION
Hutchinson-Gilford progeria syndrome is very rare disorder, less than 100 cases have been reported worldwide. Only few cases have been reported to survive after second decade. Because of its rarity, lack of reporting and long term follow up prompted us to report this rare syndrome.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Recurrent de novo point mutation in lamin A cause Hutchinson-Gilford progeria syndromeNature 2003; 423: 293-8.
2. Crounse RG, Demis DJ, Clinical Dermatology1989; I: Sixteenth Revision. Philadelphia: J.B. Lippincott Company; Unit 4 – 30, 1-5
3. Badame AJ, Review of progeriaArch Dermatol 1989; 125: 540-4.
4. Agarwal US, Sitaraman S, Mehta S, Panse G, Hutchinson-Gilford progeria syndromeIndian J Dermatol Venereol Leprol 2010; 76: 591.
5. Hamer L, Kaplan F, Fallon M, The musculoskeletal manifestations of progeria: A literature reviewOrthopedics 1988; 11: 763-9.
6. Ganesh SP, Narendra KK, Maria K, Valia RG, Valia AR, Disorders of connective tissueIADVL Textbook of Dermatology 2008; 3rd ed. Mumbai: Bhalani Publishing House; 1181-3.
7. Kanathur S, Sarvajnyamurthy S, Somaiah SA, Characteristic facies: An index of the diseaseIndian J Dermatol Venereol Leprol 2013; 79: 439-43.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.