Bullous Wells’ syndrome
Bengu Cevirgen Cemil1, Necip Enis Kaya1, Aysun Gokce2, Muzeyyen Gonul1
1Department of Dermatology, Ministry of Health Diskapi Yildirim Beyazit Education and Research Hospital, Ankara, Turkey, 2Department of Pathology, Ministry of Health Diskapi Yildirim Beyazit Education and Research Hospital, Ankara, Turkey
ABSTRACT
Wells’ syndrome (WS) is an uncommon inflammatory skin disease which typically presents single or multiple erythematous and edematous urticarial plaques similar to cellulitis. The lesions may evolve into blue-grey morphea-like lesions and may persist for weeks or months. They ultimately heal without scar. Other clinical presentations reported in literature include papular and nodular and, rarely, bullous eruptions. Previously, bullous Wells’ syndrome was rarely reported in the literature. Herein, we describe a case of a female patient with bullous Wells’ syndrome localized to the upper limbs without any associated disorders.
Key words: Bullous lesion; Drug therapy; Wells syndrome
INTRODUCTION
Wells syndrome is an acute, rare dermatosis characterized by painful and itchy urticarial and cellulitis-like plaques. The lesions evolve rapidly over 2-3 days into plaques that resolve spontaneously over 2-8 weeks without scarring. Definitive diagnosis is made by pathology which is rich edema and infiltration of eosinophils in the dermis. In the subacute stage, flame figures surrounding collagen bands are observed. The presence blisters on the lesion is very rare in Wells syndrome. Wells syndrome’s pathophysiology has not been elucidated and this disease often shows recurrence [1,2]. We present a case of Wells syndrome characterized by blisters here.
CASE REPORT
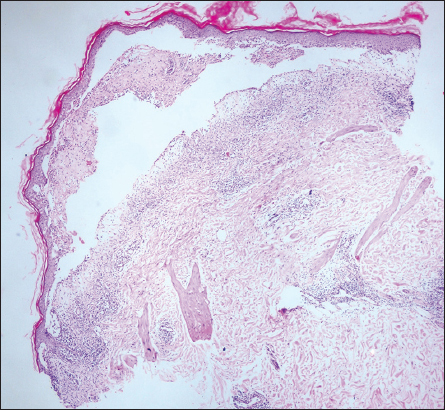
A 44-year-old female presented with redness, fluid-filled blisters and pain on left arm. Past history at both dorsum of the feet, extensor surface of the tibia and malar region redness and blisters were complaints. The patient’s resume had urticaria history and antihistamine use. On dermatological examination, an erythematous and edematous well demarcated plaque with multiple tense and flaccid bullae containing serous fluid were observed on the left forearm extensor surface (Fig. 1). The Nikolsky’s sign was negative. Blood count, peripheral smear, renal and liver function tests, immunoglobulin E (IgE) level were within normal limits. Erythrocyte sedimentation rate (ESR) in the first hour was 45 mm (0-20). Chest X-ray and ultrasonography (USG) for lymph nodes and abdominal USG produced normal results. Histopathology examination from a biopsy taken from the skin showed subepidermal blister containing, eosinophils, fibrinous material, lymphocytes and occasional polymorphonuclear leukocytes (Fig. 2). Dermal edema, perivascular, interstitial and periappenducular eosinophils intermingled with mononuclear inflammatory cells were seen in the dermis (Fig. 3). Direct immunofluorescence staining was negative. Based on the clinical presentation and the histopathology, a diagnosis of bullous Wells syndrome was made. Systemic steroid (prednisolone 60 mg/day), anti-histamine therapy (cetirizine 5 mg once daily) was combined with eau borique 2% daily dressing and topical antibiotics wound care. The lesions dramatically regressed within several days and steroid dose was tapered. The patient’s lesions healed without scarring.


Prior to the study, patient gave written consent to the examination and biopsy after having been informed about the procedure.
DISCUSSION
Classical clinic presentation of Wells syndrome is painful and slightly itchy recurrent urticarial and cellulitis-like plaques. Papulonodular lesions, bluish-green, slate gray, pink or violaseous plaques have also been described [3,4]. Blisters may occur very rarely on cellulite-like plaques. To our knowledge, bullous Wells syndrome cases have been reported less than 15 in the literature to date [5–8]. Wells syndrome affects mostly adults and may involve any race and both genders [2]. Fever, fatigue, joint pain may be seen. Blood eosinophilia and increased IgE may be found [4]. In our patient, there was no accompanying symptoms, peripheral eosinophilia and increased immunoglobulin E level.
The exact etiology of Wells syndrome are unknown, however, it has been reported that insect bites, leukemia, bacterial, viral, fungal and parasitic infections, myeloproliferative disorders, drugs may be triggering factor for the disease [1,4–6]. Our patient did not have any triggering factors. Also, the pathogenesis of the disease is not known exactly, but urticarial lesions may occur with the abnormal reaction of eosinophils [4]. Aberrant and inadequate eosinophil skin homing is one of the major fact in disease expression of Wells’ syndrome. An increase in Interleukin (IL)-5 levels have been observed due to Wells’ syndrome. IL-5 does not only mobilize eosinophils from the bone marrow but also promotes homing of eosinophils by altering expression of adhesion molecules. In addition, increased levels of IL-5 appear to induce expression of CD25, the alfa chain of the IL-2 receptor, which increases eosinophil degranulation and following tissue destruction [9].
Histopathologically, dermal edema, rich infiltration of eosinophils in the superficial and deep dermis is observed in the acute stage. Flame figures are formed in the subacute stage when degranulating eosinophils coat basophilic collagen bundles with eosinophilic major basic protein (MBP). Phagocytic histiocytes are observed around the flame figures during the resolving stage. The flame figures are distinctive, but not pathognomonic for Wells syndrome. They may occur approximately 50% of patients with Wells syndrome [3]. Also, flame figures may be seen in other dermatosis such as bullous pemphigoid, insect bite reactions, cutaneous mastocytoma, eczema, prurigo, fungal infections and herpes gestationis [1]. Flame figure was not detected in our patient. This condition may be associated with biopsy performed in the acute phase (3th days after onset of lesions).
There is bacterial cellulitis, erysipelas; arthropods bite reactions, allergic contact dermatitis, bullous pemphigoid, and Churg-Strauss syndrome in differential diagnosis of Wells syndrome. Detailed history, clinical examination and infection markers such as leukocyte count, sedimentation rate, C-reactive protein and fecal parasite search, serum IgE levels and looking parasite-specific antibodies, patch test, histopathological examination and direct immunofluorescence staining allow making differential diagnosis [10].
Topical steroids may be preferred in the treatment of mild cases, but oral corticosteroids are the first choice in severe cases. A few days therapy of systemic steroid achieved a dramatic response. Antihistamines such as cetirizine, minocycline, colchicine, antimalarials, dapsone, griserofulvin, interferon-alpha and cyclosporine are other treatment options [10]. In our case, the lesions dramatically regressed within several days with the oral cetirizine and oral prednisolone 60 mg/day therapies. Complete cure of the lesions was obtained within 2 weeks and gradually steroid therapy was discontinued. The patient has been in remission for 5 months.
In conclusion, presence of bullous lesions in the Wells’ syndrome is a rare finding. Wells syndrome should be considered at the differential diagnosis of diseases characterized by blistering.
Consent
The examination of the patient was conducted according to the Declaration of Helsinki principles.
REFERENCES
1. Karaca S, Kulac M, Aktepe F, Özel H, Eosinofilik Selülit (Wells Syndromu): Olgu SunumuKocatepe T?p Dergisi 2005; 6: 59-62.
2. Sezgin AO, Ercal HE, Karaarslan IK, Ertam ?, Dereli T, Kandillio?lu G, Eosinophilic Cellulitis: An 11-Year-Old Male PatientTurk J Dermatol 2010; 4: 40-3.
3. Bansal M, Rai T, Pandey SS, Wells syndromeIndian Dermatol Online J 2012; 3: 187-9.
4. Moossavi M, Mehregan DR, Wells’ syndrome: a clinical and histopathologic review of seven casesInt J Dermatol 2003; 42: 62-7.
5. Feliciani C, Motta A, Tortorella R, De Benedetto A, Amerio P, Tulli A, Bullous Wells syndromeJ Eur Acad Dermatol Venereol 2006; 20: 1021-2.
6. Spinelli M, Frigerio E, Cozzi A, Garutti C, Garavaglia MC, Altomare G, Bullous Wells’Syndrome Associated with Non-Hodgkin’s Lymphocytic LymphomaActa Derm Venereol 2008; 88: 530-1.
7. Katoulis AC, Bozi E, Samara M, Kalogeromitros D, Panayiotides I, Stavrianeas NG, Idiopathic bullous eosinophilic cellulitis (Wells-syndrome)J Clin Exp Dermatol Res 2009; 34: 375-6.
8. Verma P, Singal A, Sharma S, Idiopathic bullous eosinophilic cellulitis (Wells syndrome) responsive to topical tacrolimus and antihistamine combinationIndian J Dermatol Venereol Leprol 2012; 78: 378-80.
9. Gilliam AE, Bruckner AL, Howard RM, Lee BP, Wu S, Frieden IJ, Bullous “cellulitis” with eosinophilia: case report and review of Wells’ syndrome in childhoodPediatrics 2005; 116: 149-55.
10. Jean L. Bolognia, Julie V. Schaffer, Karynne O. Duncan, Christine J. Ko, Eosinophilic dermatoses. Dermatology Essentials 2014; 1st ed. Philadelphia: Saunders Elsevier; 195-8.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.