Solitary neurofibroma
Patricia Chang1, Tyson Meaux2, Gylari Calderon1
1Department of Dermatology, Hospital General de Enfermedades IGSS and Hospital Ángeles, Guatemala, 2Student, Hospital General de Enfermedades IGSS and Hospital Ángeles, Guatemala
Sir,
58 year old female with no significant past medical or family history incidentally observed to have a growth on her left fifth toe. It was soft, skin-colored, and compressible (Figs. 1 and 2). The rest of the physical examination was within normal limits.

Figure 1: Panoramic view of the lesion
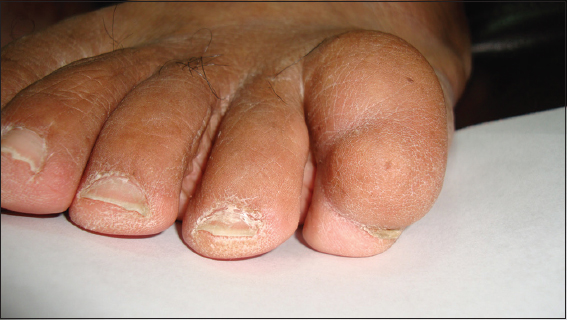
Figure 2: Closer inspection demonstrates a soft, skin-colored neoplasm spanning the length of the 5th toe
The patient reported that five years ago she noticed that the fifth left toe starting becoming “thick” and that in the last year and a half it had increased in thickness and length, covering the entire digit. She states that it does not cause discomfort and no family members have any similar lesions.
Past medical and family history: negative.
With this clinical data, the diagnosis of a soft tissue tumor was made and the patient underwent biopsy of the lesion.
Biopsy of the lesion at scanning power shows laminar hyperkeratosis of the epidermis, slight irregular acanthosis, and a perivascular lymphocytic inflammatory infiltrate in the papillary and superficial reticular dermis (Fig. 3), in the mid reticular dermis there is an unencapsulated, poorly defined neoplasm, consisting of a symmetrically arranged proliferation of wavy spindle cells in fascicles, immersed in a fibromyxoid matrix (Fig. 4) also observed are nerve bundles which surround the subcutaneous tissue and are indeed the origin of the lesión. Higher magnification demonstrates an abundant fibromyxoid stroma, with unorganized spindle cells with wavy nuclei, basophils, and indistinct cytoplasmic borders (Fig. 5). Higher magnification shows that the wavy spindle cells are actually Schwann cells and peri-neural fibroblasts, interspersed with mast cells and axons in a myxoid stroma (Fig. 6).
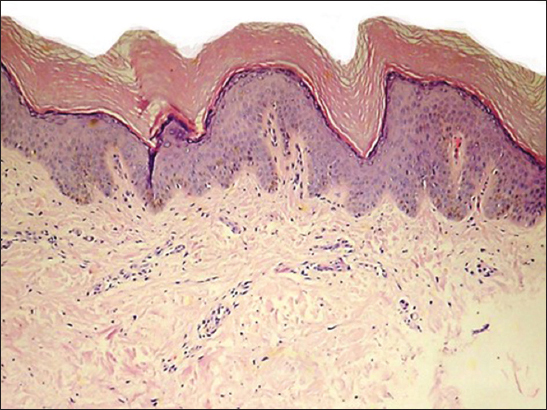
Figure 3: At scanning power, there is laminar hyperkeratosis of the epidermis, slight irregular acantosis, and a perivascular lymphocytic inflammatory infiltrate in the papillary and superficial reticular dermis
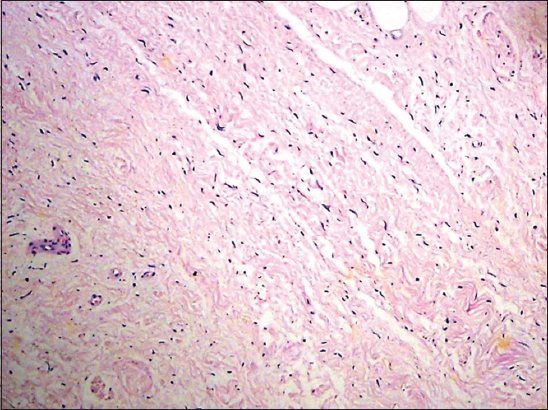
Figure 4: In the mid reticular dermis is an uncapsulated poorly defined neoplasm consisting of a symmetrically arranged proliferation of wavy spindle cells in fascicles, immersed in a fibromyxoid matrix

Figure 5: Nerve bundles which surround the subcutaneous tissue and are indeed the origin of the lesión. Higher magnification demonstrates an abundant fibromyxoid stroma, with unorganized spindle cells with wavy nuclei, basophils, and indistinct cytoplasmic borders
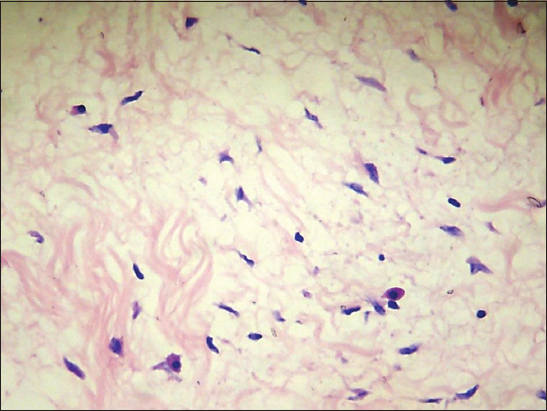
Figure 6: Higher magnification shows that the wavy spindle cells are actually Schwann cells and peri-neural fibroblasts, interspersed with mast cells and axons in a myxoid stroma
Prior to the study, patient gave written consent to the examination and biopsy after having been informed about the procedure.
This clinical case is interesting because it is a solitary neurofibroma without neurofibromatosis.
Neurofibromas are soft, rubbery, pink to skin-colored benign tumors of the peripheral nerve sheath that invaginate when central pressure is applied, known as the “buttonhole” sign [1–3]. Solitary neurofibromas typically arise in the second or third decade of life as asymptomatic, slowly-enlarging, soft growths [4]. They may present as papules or nodules, become pedunculated over time, and more than one is present in 10% of cases. They are most commonly found on the skin of the head and neck [5]. The presence of one or two solitary neurofibromas is not usually a cause for concern, however, the diagnosis of neurofibromatosis should be considered if three or more are present. On the other hand, one plexiform neurofibroma, which mostly occurs on the trunk and proximal extremities and presents as an occasionally pigmented, bag-like mass, is pathognomonic for Neurofibromatosis 1. Neurofibromatosis types 1 and 2 are autosomal-dominantly inherited neurocutaneous disorders with a significantly increased risk of tumors in various organs.
Type 1 is more common of the two, mainly affecting the skin, skeletal, and peripheral nervous systems. Neurofibromatosis 2 on the other hand, is a very rare disorder with a low incidence of skin manifestations and a classic association with bilateral acoustic neuromas [3,6]. On biopsy, solitary neurofibromas demonstrate wavy, spindled nuclei, fine collagen fibers, and a myxoid stroma with an abundance of mast cells. Histological appearance varies according to the amount of mucin and myxoid tissue present. Cholinesterase activity, S-100, vimentin, and myelin basic protein are positive markers. Histologically and clinically, neurofibromas are identical in behavior, regardless of whether or not they are occurring as part of neurofibromatosis [7,8]. Clinically, 90% manifests as solitary, soft, small, skin-colored, neoplasms, occasionally exceeding 6 cm in size [8]. While the clinical differential diagnosis includes dermal melanocytic nevus, schawannoma, soft fibroma, and fibrolipoma, the diagnosis of neurofibroma should be considered for all slow-growing, soft or rubbery swellings on the skin, regardless of the location [2,9]. Interestingly, there have been a handful of case reports of subungual neurofibromas, in which the differential diagnosis includes glomus tumor. Glomus tumors, however, can be distinguished from neurofibromas by the symptoms of hypersensitivity to cold, paroxysmal severe pain, and point tenderness in the involved finger or toe [10]. In addition to the skin, numerous cases of head and neck neurofibromas have been reported, including one report in the palatine tonsil. While solitary cutaneous neurofibromas are almost always benign, those of the head and neck are typically deep-seated, and have a 5-12% chance of malignant transformation [11]. Treatment of neurofibromas is indicated if symptomatic, and generally consists of surgical excision [1]. However, if the tumor is soft and small, better cosmesis may be obtained by extruding the tumor through a small punch hole [2].
CONSENT
The examination of the patient was conducted according to the Declaration of Helsinki principles. Written informed consent was obtained from the patient for publication of this article.
REFERENCES
1. James WD, Berger TG, Elston DM, Dermal and Subcutaneous Tumors in: Andrews’ Diseases of the Skin: Clinical DermatologySaunders Elsevier 2006; 609
2. Prasad-Hunasehally RY, Motley R, Bulbous swelling on the finger pulpClinical and Experimental Dermatology 2009; 34: 279-80.
3. Vargas EC, Nole KB, Chávez SM, Neurofibroma circunscrito solitarioDermatol Perú 2008; 18: 55-8.
4. Venadero-Albarrán F, Rodríguez AM, Merelo V, Cervantes AAM, Ramos-Garibay A, Neurofibroma solitario. Comunicación de dos casosRev Cent Dermatol Pascua 2004; 13: 121-3.
5. Argenyi ZB, Neural and Neuroendocrine Neoplasms (Other Than Neurofibromatosis) in: Bologna, DermatologySaunders Elsevier 2012; 1949
6. Ferner RE, Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspectiveLancet 2007; 6: 340-51.
7. Oshman RG, Phelps RG, Kantor I, A Solitary Neurofibroma on the FingerArch Dermatol 1988; 124: 1185-86.
8. Flórez L, Barajas JS, Neurofibroma solitario en el paciente sin neurofibromatosis: Aspectos biológicos y clínicosMedUNAB 2008; 11: 61-5.
9. Argenyi ZB, Neural and Neuroendocrine Neoplasms (other than Neurofibromatosis) in: Dermatology2012; 3rd ed. Elsevier Limited; 1950
10. Huajun J, Wei Q, Ming L, Chongyang F, Weiguo Z, Decheng L, Solitary subungual neurofibroma in the right first fingerInt J Dermatol 2012; 51: 335-8.
11. Madhumita K, Nambiar A, Prathapan P, Solitary neurofibroma of the palatine tonsil: A case reportEar Nose Throat J 2007; 86: 756-8.
Notes
Source of Support: Nil,
Conflict of Interest: None declared.
Comments are closed.